|
Iron Sulfur
Clusters
We have been fortunate to be involved in characterizing novel structures
such as the nitrogenase M and P-centers,1 the coupled
heme-[4Fe-4S] chromophore of E. coli. sulfite reductase,2
[3Fe-4S] clusters,3 the clusters of carbon monoxide
dehydrogenase,4 the key FeIVFeIV
intermediate (compound Q) in the catalytic cycle of methane
monooxygenase,5 the H-cluster of [Fe]-hydrogenases,6
and the oxygen sensor of E. coli.7 For well established
structures, we have been interested in attaining new oxidation states, e.g.
the all-ferrous [4Fe-4S] cluster.8 In general terms, our
interests are described in Ref 9. We continue to study a variety of
iron-sulfur proteins.
|
|

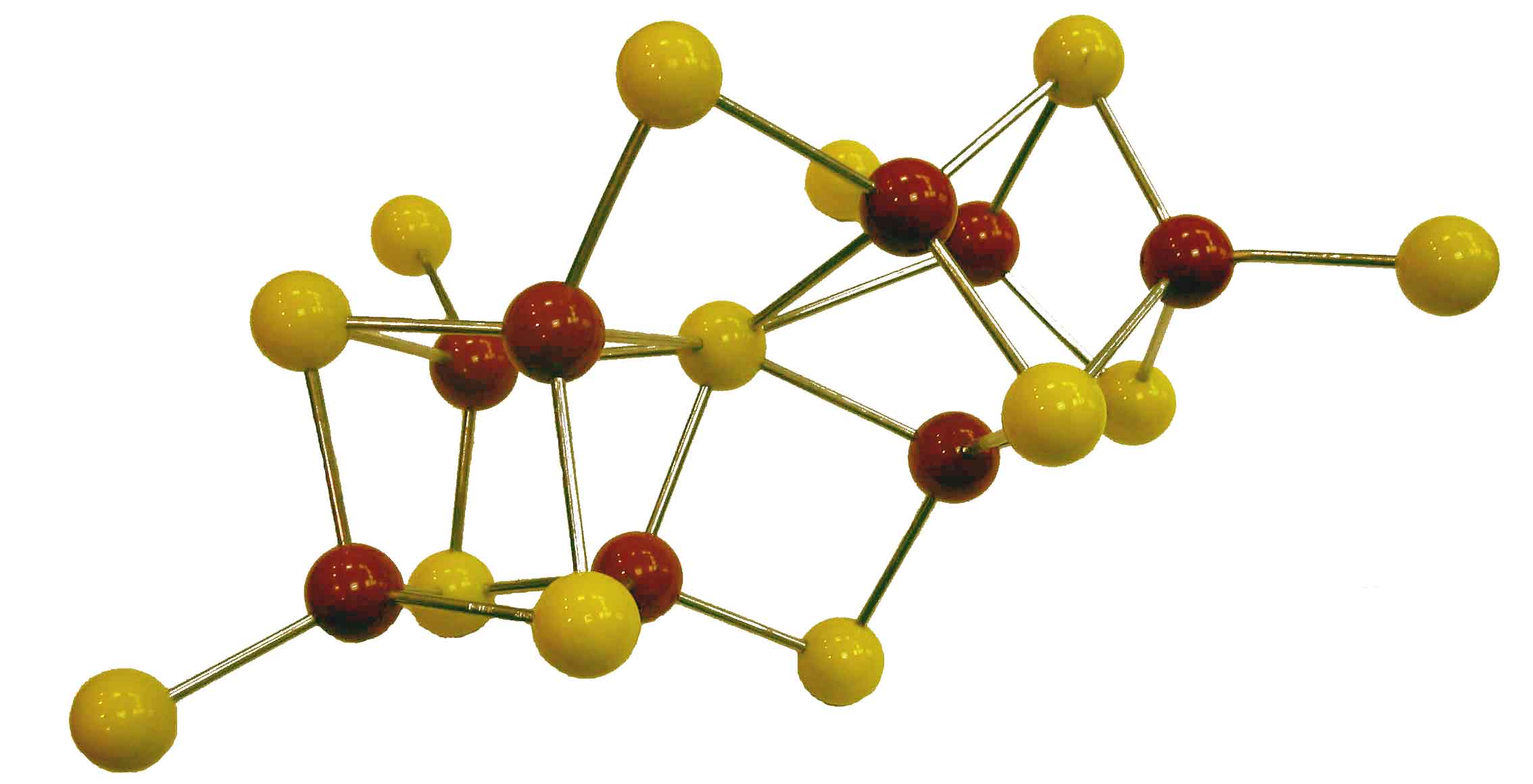
Our idea of the nitrogenase
P-cluster shortly
before crystallographic refinement by Rees et al. (1992)
|
|
(1) Surerus, K. K.;
Hendrich, M. P.; Christie, P.; Rottgardt, D.; Orme-Johnson, W. H.; Münck,
E. J. Am. Chem. Soc. 1992, 114, 8579-8590.
(2) Bominaar, E. L.; Hu, Z.; Münck, E; Girerd,
J.-J.; Borshch, S. J. Am. Chem. Soc. 1995, 117,
6976-89.
(3) Emptage, M. H.; Kent, T. A.; Huynh, B.
H.; Rawlings, J.; Orme-Johnson, W.H.; Münck, E. J. Biol. Chem. 1980,
255, 1793-1796.
(4) Xia, J.; Hu, Z.; Popescu, C. V.; Lindahl,
P. A.; Münck, E. J. Am. Chem. Soc . 1997, 119,
8301-12.
(5) Shu, L.; Nesheim, J. C.; Kauffmann, K.;
Münck, E.; Lipscomb, J. D.; Que Jr., L. Science 1997,
275, 515-518.
(6) Popescu, C. V.; Münck, E. J. Am.
Chem. Soc. 1999, 121, 7877-7884.
(7) Popescu, C.; Bates, D. M.; Beinert, H.;
Münck, E.; Kiley, P. J. Proc. Natl. Acad. Sci. USA 1998,
95, 13431-13435.
(8) Yoo, S. J.; Angove H. C.; Burgess, B.K.;
Hendrich, M.P.; Münck, E. J. Am. Chem. Soc. 1999,
121, 2534-2545.
(9) Beinert, H.; Holm, R.H.; Münck, E.
"Iron-Sulfur Clusters: Nature’s Modular Multipurpose Structures" Science
1997, 277, 653-659.
(10) Chakrabati, M.; Deng, L.; Holm, R. H.; Munck, E.;
Bominaar, E. L. Inorg. Chem. 2009, 48, 2735-2727
(Cover page Inorg. Chem. Apr. 6, 2009).
|
|
High-Valent
complexes of Biological Relevance
Together
with the group of Lawrence
Que, Jr., at the University of Minnesota, we are studying the
electronic structures of high-valent iron complexes relevant to oxygen
activation. Our joint projects have produced a larger number of interesting
compounds. Among these is the first nonheme FeIV-oxo complex,1
[FeIV(O)(TMC)(NCMe)]2+. This complex has
electronic spin S = 1. For nonheme proteins the combination of carboxylate,
histidine, H2O, OH- ligands generally produces
high-spin (S = 2) FeIV sites, an environment not easily produced
in a synthetic complex.
Collaborating with the groups of
T. J. Collins here at CMU and L. Que, Jr. we reported in 2008 a comprehensive
spectroscopic study of the first FeV=O complex, using the
Collins TAML ligand (figure 2).2 In
2012 we followed with the second FeV=O complex.3 This
complex was generated by reacting [FeIV(O)(TMC)(NCMe)]2+
at -44 °C with tert-butyl hydroperoxide in the presence of a strong base.
The new species exhibits in the glass-forming 3:1 butyronitrile/MeCN
solvent an S = 1/2 EPR signal with very narrow lines (4 gauss). The
high-resolution allowed mapping of 14N, 57Fe and
17O hyperfine tensors by EPR. It required detailed DFT analysis
(with choice of a suitable functional) to interpret the Mossbauer, EPR and
resonance Raman data in a consistent way. The S = 1/2 species turned out to
be an iron(V) complex having axial oxo and acetylimido ligands, namely [FeV(O)(TMC)(NC(O)CH3)]+.
We are currently pursuing other systems for which FeV=O species
are indicated, including complexes implicated in water oxidation.
We have
recently studied a group of high-valent diiron complexes based on the TPA
ligand (traded by insiders as the Castro Brothers). The FeIVFeIV
complex has two local S = 1 sites which are ferromagnetically coupled to
yield an S = 2 system state.4 One site, Feb, has a
terminal oxo group; Fea has a hydroxo ligand. (figure 3) Given
that the Fe-O-Fe angle is 130°, the observation of ferromagnetic coupling
was puzzling, but could be explained quite well after realizing that the
two sites have different ligand fields that produce a crucial pair of
orthogonal magnetic orbitals. Reduction by one electron renders Fea
site high-spin FeIII. Concomitantly Feb undergoes a
transition to high-spin (Sb=2) FeIV=O, a
transition that is driven by superexchange interactions between Fea
and Feb (By going high-spin, the FeIV=O
site enables efficient antiferromagnetic pathways between the two Fe).5
For the same TPA ligand our collaborators have produced dinuclear complexes
with open and closed cores, such as O=FeIV-O-FeIV-OH
and FeIV(µ-O)2FeIV. Core opening increases
H-bond cleaving reactivity roughly 1000-fold; the spin transition at the
oxo site yields an additional 1000-fold increase (see Fig. 4 of ref 6).
|
|
|

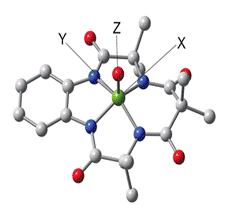
[FeIV(O)(TMC)(NCMe)]2+ (TAML)FeV=O
|
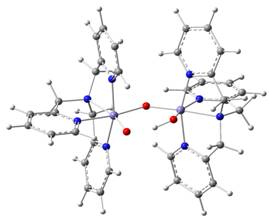
(TPA)(O)FeIV-O-FeIV(OH)(TPA)
|
|
|
|
(1)
Rohde, J.-U.; In, J.-H.; Lim, M. H.; Brennessel, W. W.; Bukowsky, M. R.;
Stubna, A.; Münck, E.; Nam, W.; Que Jr., L. Science 2003,
299, 1037-1039.
(2) Tiago
de Oliveira, F.; Chanda, A.; Banerjee, D.; Mondal, D.; Bominaar, E.; Münck,
E.; Collins, T. C. Science 2006, 315, 835-838. (PMID:
17185561)
(3) Van
Heuvelen, K. M.; Fiedler, A. T.; Shan, X.; De Hont, R. F.; Meier, K.
K.; Bominaar, E. L.; Münck, E.; Que, Jr., L. Proc. Natl.
Acad. Sci .2012, 109, 1193-38.
(4)
Martinho, M.; Xue, G.; Fiedler, A. T.; Que, Jr., L.; Bominaar, E. L.;
Münck, E. J. Am. Chem. Soc. 2009, 131, 5823-5830.
(5) De
Hont, R. F.; Xue, G.; Hendrich, M. P.; Que, L. Jr.; Bominaar, E. L.; Munck,
E. Inorg. Chem. 2010, 49, 8310-8322.
(6) Xue,
G.; De Hont, R. F.; Münck, E.; Que, L. Jr. Nature Chem. 2010,
2, 400-405.
|
|
|
|
|
|
|
|
Dioxygenases
and Monooxygenases
Together with the group of J. D. Lipscomb at the University of Minnesota
we are studying various oxygen activating enzymes, among them the Fe2+
homoprotocatechuate 2,3 dioxygenase (2,3 HPCD) which cleaves the aromatic
ring of homoprotocatechate (HPCA) adjacent to the vicinal hydroxyl groups.
This enzyme represents a large group of dioxygenases involved in bacterial
degradation pathways of natural and 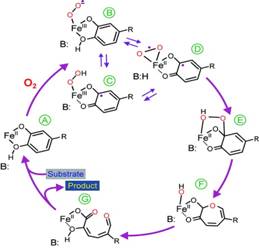 man-made compounds. In the native
state of 2,3 HPCD the Fe2+ is coordinated by a 2-His-1-Glu
facial triad; the three remaining coordination sites are occupied by water
molecules which are displaced upon (bidentate) substrate and O2
binding. The figure summarizes current ideas of the mechanism. After oxygen
binds, an electron is transferred through the iron to the oxygen, giving
both substrates radical character (SQ·
-FeII-O2·-
in step D ). Recombination of the radicals would yield an
alkylperoxo intermediate, step E. A man-made compounds. In the native
state of 2,3 HPCD the Fe2+ is coordinated by a 2-His-1-Glu
facial triad; the three remaining coordination sites are occupied by water
molecules which are displaced upon (bidentate) substrate and O2
binding. The figure summarizes current ideas of the mechanism. After oxygen
binds, an electron is transferred through the iron to the oxygen, giving
both substrates radical character (SQ·
-FeII-O2·-
in step D ). Recombination of the radicals would yield an
alkylperoxo intermediate, step E. A 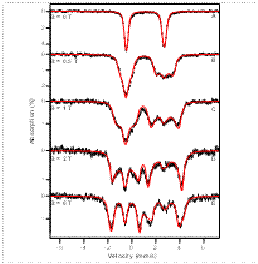 subsequent
Criegee-type rearrangement would result in O-O bond cleaving to yield a
lactone intermediate, with the second oxygen retained on the iron. Hydrolysis
of the lactone by this oxygen would then yield the product. subsequent
Criegee-type rearrangement would result in O-O bond cleaving to yield a
lactone intermediate, with the second oxygen retained on the iron. Hydrolysis
of the lactone by this oxygen would then yield the product.
For the native
enzyme and three mutants we have followed the catalytic reaction with rapid
freeze-quench Mössbauer and EPR spectroscopy and characterized a variety of
intermediates, among them an Fe3+-superoxo species and a
semiquinone-Fe3+-hydroperoxide complex (1-3). For
native HPCD and its mutants, E. Kovaleva and J. D. Lipscomb have
obtained > 50 high-resolution X-structures as well as a wealth of
kinetic data. For a variety of states we have obtained well resolved
Mössbauer spectra which, in conjunction with quantum chemical calculations,
can be used to gain insight into the electronic structures of crucial
intermediates. This will allow us to relate electronic and X-ray structure
data along the reaction pathways and thus obtain crucial information about
reactivity. The figure shows Mössbauer spectra of an E-S complex; red
lines: spectral simulations).
We are continuing
ongoing studies aimed at characterizing intermediates of the reaction
cycle of methane monooxygenase, such as species Q, Q' and P*.
(1)
Mbughuni, M. M.,
Chakrabarti, M., Hayden, J. A., Bominaar, E. L., Hendrich, M. P., Münck,
E., and Lipscomb, J. D. (2010) Trapping and spectroscopic characterization
of an FeIII-superoxo intermediate from a nonheme mononuclear
iron-containing enzyme. Proc. Natl. Acad. Sci. U. S. A. 107,
16788-16793.
(2)
Mbughuni, M. M.,
Chakrabarti, M., Hayden, J. A., Meier, K. K., Dalluge, J. J., Hendrich, M.
P., Münck, E., and Lipscomb, J. D. (2011) Oxy-intermediates of
homoprotocatechuate 2,3-dioxygenase: Facile electron transfer between
substrates. Biochemistry 50, 10262-10274.
(3)
Mbughuni, M. M.;
Meier, K. K.; Münck, E.; Lipscomb, J. D. "Substrate-Mediated Oxygen
Activation by Homoprotocatechuate 2,3-Dioxygenase: Intermediates Formed by
a Tyrosine 257 Variant" Biochemistry 2012,51,
8743-54.
Electronic
Structure Analysis
The spectroscopic studies of our group have often been complemented by DFT
calculations. These computations provide theoretical estimates of
experimentally determined spin-Hamiltonian parameters, such as zero-field
splittings, exchange-coupling constants, 57Fe isomer shifts,
quadrupole splittings, and magnetic hyperfine coupling constants, and give
detailed insights into the dependency of these parameters on molecular
geometry and electronic structure. These studies have clarified the origin
of the unquenched orbital momentum in the diketiminate complexes of iron,1,2
the intrinsic mechanism for the distortion of the Fe(SR)4 center
in rubredoxins,3 the influence of excited spin triplet states on
the zero-field splitting in reduced form of these centers,4 and
the oxidation state of the cofactor in nitrogenase.5
(1)
Andres, H.; Bominaar, E.L.; Smith, J.M.; Eckert, N.A.; Holland, P.L.;
Münck, E. J. Am. Chem. Soc. 2002, 124, 3012-3025.
(2) Stoian, S.A.; Yu, Y.; Smith, J.M.; Holland, P.L.;
Bominaar, E.L.; Münck, E. Inorg. Chem. 2005, 44,
4915-4922.
(3) Vrajmasu, V.V.; Münck, E.; Bominaar, E.L. Inorg. Chem.
2004, 43, 4862-4866; ibid. 4867-4879.
(4) Vrajmasu, V.V.; Bominaar, E.L.; Meyer, J.; Münck, E. Inorg.
Chem. 2002, 41, 6358-6371.
(5) Vrajmasu, V.V.; Münck, E.; Bominaar, E.L. Inorg.
Chem. 2003, 42, 5974-5988.
|
|