| Cerius2 Modeling Environment |
This chapter is divided into three parts:
In this tutorial, you familiarize yourself with the look and feel of the Cerius2 GUI and Visualizer. You use the Cerius2 menu bar (File, Edit, Build, etc.) and the tool bar immediately below it. You learn how to load a model and then practice viewing, displaying, and selecting it. The tutorial concludes with a brief exercise about control panels.
Summary of tutorial
The tutorial is divided into four short exercises:
1. Start Cerius2 and load the model.
If Cerius2 isn't already open, type
> cerius2in a UNIX shell to run the program. |
| If typing cerius2 does not start the program, consult the Cerius2 Installation and Administration Guide for help on setting up the correct environment for running Cerius2. |
| Select File/Load Model... to open the Load Model control panel. |
Remember to use the left mouse button for choosing menu items.
| You may need to use the scroll bar at the side of the file list to locate the organics/ directory. |
Your model window should look similar to this:
2. Change the display style of the model.
| Use the Display Style popup menu on the left end of the tool bar to change the display style to Ball. |
The atoms are displayed as spheres. By default they are filled to 0.7 of their van der Waals radii.
| Select Stick from the Display Style popup menu. |
The model returns to stick display where all bonds drawn as lines.
B. Viewing and labeling a model
The following steps show you how to rotate and translate the trimethylbenzene model. You also learn how to label atoms in the model by element name.
| Rotate the model by placing the cursor over the model window and then dragging while holding down the right mouse button. Familiarize yourself with the action of the mouse. |
Note that rotation is quickest in the stick display.
| Change the Atom Labeling popup on the tool bar from No Label to Elements. |
All the atoms in the model window should now be labeled as hydrogens or carbons, either C or H.
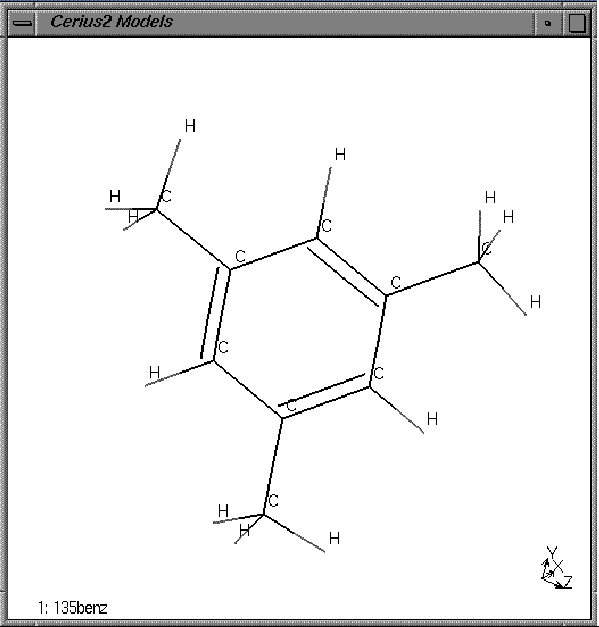
C. Selecting atoms
In the following steps, you learn several techniques for selecting and deselecting atoms.
| Use the left mouse button to pick one of the atoms in the model. |
| Select several more atoms by holding down the <Shift> key as you select them. |
You should now have three or four atoms highlighted in the model window.
| Use the Display Style popup to change to Ball & Stick. |
Note that the new display style only applies to the selected atoms. In the ball and the cylinder display styles, selected atoms are indicated with thin stripes.
| Deselect all atoms by clicking on empty space in the model window. |
|
Find the Select All and Deselect All |
Next, you use these buttons to select and deselect atoms in the model window.
| Choose the Select All button then changethe Display Style popup to Ball. |
All the atoms in the window are highlighted and then appear as balls wrapped in thin blue lines.
| Choose the Deselect All button. |
All the atoms are deselected -- the thin blue lines disappear.
D. Manipulating control panels
In the following steps, you learn about control panels: how to open, close, scale, and stack them. You use on-line help to get information about the controls on the panels. And finally you learn how to re-initialize and/or exit from Cerius2.
|
Close the Load Model control panel by clicking on the close box |
All control panels can be closed this way.
| Open the Save Model control panel by choosing File/Save Model... . |
Notice that those pulldown menu items that open control panels all have names that end in ellipses (...).
| Choose Build/Element Defaults to open the Edit Elements control panel. |
| Choose View/Display Attributes... to open the Display Attributes control panel. |
The Display Attributes control panel has the same functionality as the Display Style and Atom Labeling tool bar popups, which you have already used.
| Click the Preferences... button on the Display Attributes control panel to open the Style Preferences control panel. |
Again, control panel buttons with names ending in ellipses (...) open other control panels.
Click with the right mouse button on a command in one of the control panels. 
|
This brings up a help text panel that describes the function.
| Click in the help text panel to close it. |
Try clicking with the right mouse button in other items in the GUI: check boxes, buttons, popups, sliders, menu items, and so on.
| Select View/Graphics> and then move the mouse to the right to select the Stereo... item on the pullright menu. |
The Stereo control panel opens. You would use the Stereo view control panel if you wanted to view the model as a stereo pair or with the Crystaleyes stereo hardware.
| Click and drag some control panel title bars to different screen locations. |
|
Resize some control panels by clicking and dragging their edges or corners.
Click the Cascade Panels button on the tool bar. |
This stacks all the open control panels with their title bars visible.
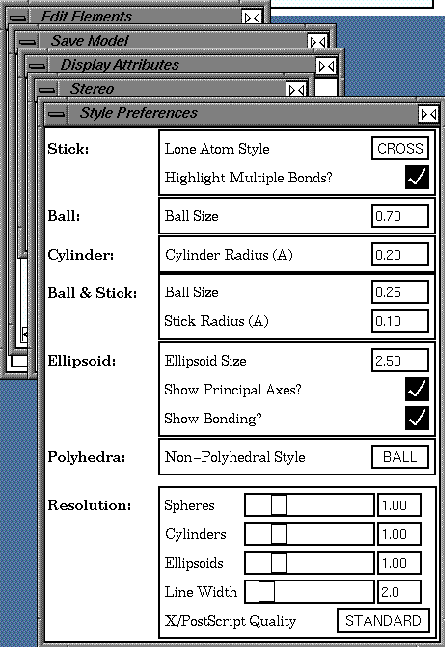
| Bring the last panel to the front by clicking on its title bar, and close it by selecting its close box (in the top right corner). |
|
Select the Clear Panels button on the tool bar. |
The remaining control panels close.
| Re-initialize the program by selecting File/New Session and selecting the Confirm button on the Re-initialize dialog box. |
Re-initialization resets all the Cerius2 control panels to their original defaults.
| Choose File/Exit to quit from Cerius2, and select the Exit button on the Exit Option... dialog box. |
Review
In this tutorial you learned how to:
Alternatively, you may want to learn more about the Visualizer by reading the user manual sections listed below.
Related material
The Visualizer is discussed in detail in the Cerius2 Modeling Environment. The sections listed here will provide you with more detailed information about atom display and selection than are included in this tutorial book. 1. Sections in the Introducing Cerius2 and Viewing and Displaying
Models chapters of the Cerius2 Modeling Environment.
3. The Managing Models and Sessions chapter of the Cerius2 Modeling
Environment.
This chapter contains a tutorial on molecular sketching. It introduces the Cerius2 3D Sketcher. You learn how to draw and edit models in 3D and how to use the Clean function to do simple optimization of model geometry. You also learn how to save the models, which you build, into structure files. ![]()
Using the 3D Sketcher
Before you begin the tutorial on page 50, you should be familiar with the contents of the section Basic GUI Functions. If you have not read this section, you should ensure that you know how to do the review items listed in the Review on page 61 before beginning this tutorial.
In Basic GUI Functions, you loaded a model from file into the model window. The purpose of this tutorial chapter is to show you how you can build your own models using the 3D molecular Sketcher. The Sketcher is the principal building tool of the Cerius2 Visualizer.
Below is an illustration of the Sketcher control panel. The Sketcher provides tools for mouse-driven sketching, editing, and cleaning of models. The operation of the Sketcher is modal, that is, the function of the left mouse button in the model window is determined by which icon is currently selected in the Sketcher control panel. The default setting is to have the Selection mode icon highlighted. This allows normal Cerius2 operation, with the mouse button selecting atoms (just as in the previous tutorial). It is a good habit to always highlight the Selection mode icon before closing the Sketcher control panel.
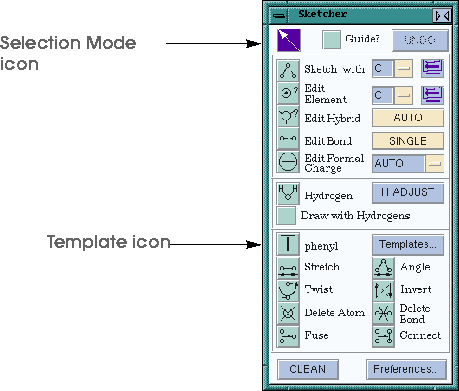
The tutorial is divided into five short exercises:
1. Start Cerius2 and open the Sketcher.
If Cerius2 isn't already open, type
> cerius2in a UNIX shell to run the program. |
| Open the Sketcher control panel by selecting the Build/3D-Sketcher... item. |
The Sketcher control panel appears.
|
Choose the Sketch icon: |
| Click once in the model window. |
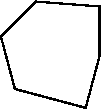
| Click somewhere else to put down a second atom and continue to build a ring of six carbon atoms --terminate the ring by clicking on the original atom. |
You should now have a hexagonal form somewhat similar to this in the model window:
| Highlight the Selection mode icon once more so that all future clicks in the Model window do not result in new atoms! |
4. Adjust the hydrogens and minimize the structure.
| Click the H Adjust button. |
| Hold down the Clean button. Release the button when the atoms stop moving. |
Clean attempts to adjust bond lengths and angles to reasonable values. Note that recleaning after manipulating or deforming the structure may find a different structure.
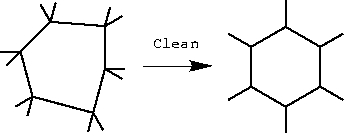
| Try altering display styles and rotating and rescaling the model to view it. (If you need hints on how to do this, refer to the Basic GUI Functions tutorial lesson.) |
|
Reset the view -- use the |
6. Save the cyclohexane model into a structure file.
B. Converting cyclohexane into the boat conformation
In the following steps, you learn how to move atoms to convert the chair conformer of cyclohexane into the boat conformer.
| Use the mouse to rotate the model to view the "chair" from the side. |
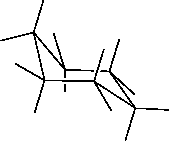
| Select the carbon on one end of the ring as you look at it, and select the two hydrogens attached to that carbon. |
(Remember you need to hold down the <Shift> key to select multiple atoms.)
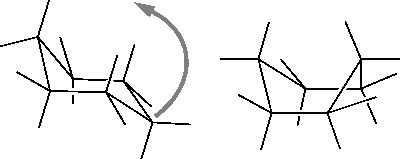
| Clean the model. |
| If cleaning returns the model back to the chair form, drag the CH2 group even farther above the seat of the chair than it was in step 3 and even closer to the back of the chair. Then repeat the Clean step. |
| Rotate and view the model. |
C. Editing cyclohexane to make benzene
In the following steps, you learn how to edit bonds and to edit and delete atoms, and so build benzene from cyclohexane.
| Find the two popups on the tool bar that currently read Atom. Change the second of these to El for element. |
| Select one hydrogen atom. |
All hydrogens appear selected in the model window.
| Double-click the Delete Atom icon in the Sketcher control panel. |
All the hydrogens in the model are deleted.
| Change the second selection rule popup back to its default Atom setting. Also, highlight the Selection mode icon once more. |
| Change the Edit Bond popup from Single to Double and click any carbon atom. Pick an adjoining carbon atom. |
A dotted guideline appears and then becomes the double bond between the carbon atoms.
| Repeat to make alternate bonds in the ring double. |
3. Adjust the hydrogens and minimize the model.
| Use the H Adjust and Clean buttons, then rotate and view this planar benzene. |
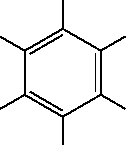
D. Editing benzene to make phenol
In the following steps, you add an OH group to benzene to make phenol. Also, you learn how to use the Sketcher tools to alter torsion angles, bond angles, and bond lengths.
| Use the popup adjacent to the Edit Element text box to select O for oxygen. |
The popup is a fast way to choose commonly used element symbols.
| Select the periodic table icon after this popup. |
| Select any element in the table. |
The element is written to the text box.
| Close the periodic table. If necessary, restore the Edit Element symbol to O. |
| Pick any one of the hydrogens to change it to an oxygen. |
In Edit Element mode, the mouse cursor is a circle.
| Use the H Adjust and Clean buttons. Highlight the Selection mode icon once more. Rotate and view the model; try some other display styles, then return to the Stick display. Choose the Twist icon. |
In the model window, the mouse cursor for twist mode is shaped like a bow tie.
| Pick the hydrogen attached to the oxygen, the oxygen itself, the carbon attached to the oxygen, and one of the adjoining ring carbons. |
| Drag the mouse over the model window with the left mouse button depressed to increase/decrease the torsion. |
The change in the torsion angle is interactively updated. Note that the atom picked last is the one that moves.
| Use the Stretch and Angle tools, which work similarly to Twist. |
| Save the phenol structure into a file called phenol.msi. If necessary, see Step 6 on page 68 for a reminder of how to save. |
| Clear the model window. |
E. Building xylene from template fragments
In the following steps, you use the template tool to create a model of p-xylene. The template tools let you sketch with predefined molecular fragments (called templates) -- for common structures, this is often more convenient than sketching atom-by-atom.
|
Click the Template icon on the Sketcher -- this looks like a "T" and is located about halfway down the box. |
The current template is phenyl.
| Click in the model window. |
A phenyl ring appears.
| Click the Templates... button in the Sketcher. Double-click the organic/ directory and find the template file methyl. Double-click methyl. |
| Pick one of the phenyl hydrogens. |
The hydrogen atom is replaced by a methyl group to make toluene.
| Select another of the phenyl hydrogens -- choose the one farthest away from the methyl group to make p-xylene. |
| Save the p-xylene structure into a file called xylene.msi. If necessary, see Step 6 on page 68 for a reminder of how to save. |
5. If you are going to continue immediately with another tutorial:
Re-initialize the program by selecting File/New Session and selecting the Confirm button on the Re-initialize dialog box.
Re-initialization resets all the Cerius2 control panels to their original defaults.
| Choose File/Exit to quit from Cerius2 and select the Exit button on the Exit Option... dialog box. |
Review
In this tutorial you learned how to ...
This chapter contains a progressive tutorial designed to show you some more functions of the Visualizer including managing multiple models, copying between model spaces, measuring models, and printing. You also get additional practice at loading, saving, and sketching models. ![]()
Managing Models and More
Before you start this tutorial lesson (page 50), you should have read (or be familiar with the contents of) Basic GUI Functions, and have done the tutorial exercises in Using the 3D Sketcher.
In this tutorial, you load in and use the models that you created in Using the 3D Sketcher.
The tutorial is divided into five short exercises:
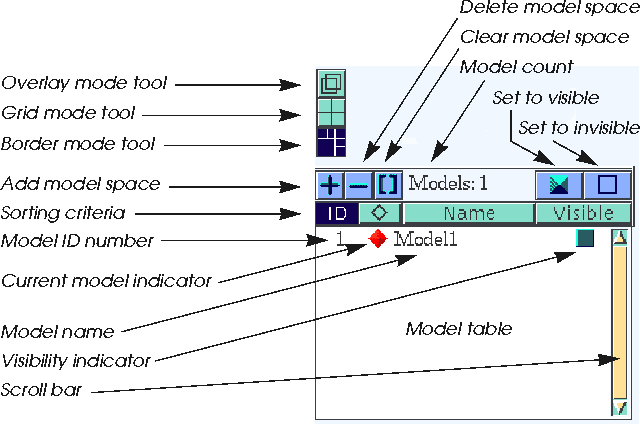
1. Load the models.
| If Cerius2 isn't already running, type cerius2 in a UNIX shell to start the program. |
| Choose Load Model... from the File pulldown menu. Use the Load Model control panel to load models from the previous tutorials: phenol.msi, cyclohexane.msi, and xylene.msi. |
| Load butane.msi and napthalene.msi from the -Cerius2-Models/organics directory. |
| Click the diamond for the different models in the list. After trying the other models, select phenol to be displayed. |
The model displayed in the window changes with each selection. Phenol should be the last one displayed.
| Check the Model Display Selector box for xylene. |
| Select two more models. |
The four models are displayed in border mode, with the current model (phenol) as a large model surrounded by smaller images of the other models.
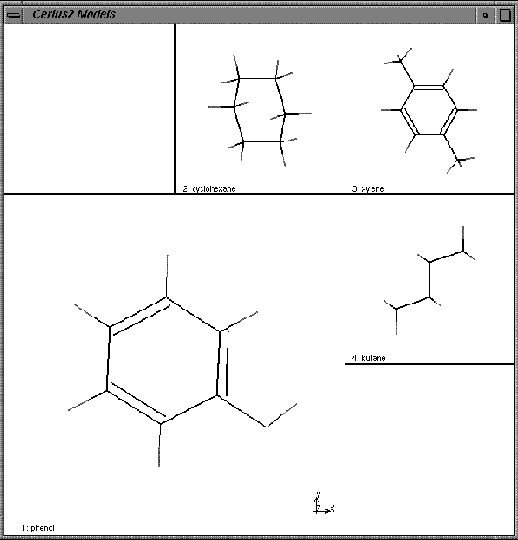
| Use the mouse to rotate and translate the models in various ways. |
Notice how all are rotated together.
| Click on the middle Display Mode icon to get a grid display. |
| Click the top icon. |
It overlays all currently visible models.
| Return to border display mode by clicking the bottom icon. Click the filled blue box on the bar above the Model Manager list. |
All models should now be displayed.
| Click the adjacent empty box icon. |
Only the current model is displayed.
| Click the Name button immediately above the list. |
| Try clicking on the ID, A, and Visible buttons to sort models in other ways. |

| Make napthalene the current model. Click the "-" icon above the Model Manager list and choose the OK button when prompted by the dialog box. |
| Make cyclohexane the current model. Click the "[ ]" icon and choose the OK button when prompted by the dialog box. |
This clears the model -- leaving it in the list but removing all data from it.
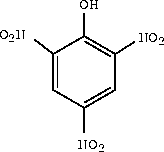
B. Making picric acid from phenol (transferring data between model spaces)
In the following steps, you learn how to create a new model space and to copy atoms from one model space and paste them into another. You copy the phenol model into the new model space and then use the Sketcher to modify the phenol model to form picric acid (2,4,6-trinitrophenol).
| Set the current model to be phenol. Select all the atoms in the phenol model. |
| Choose the Copy option from the Edit pulldown. |
This copies the phenol model into the buffer.
| Choose the "+" icon on the Model Manager. |
A new model space, called Model6, is created.
| Choose Paste from the Edit menu. |
The phenol model is copied into the new model space.
| Open the Sketcher, and click the Fragments button. Load the nitro template -- the templates/organic/nitro file. Use the nitro template to create 2,4,6-trinitrophenol. |
This molecule, 2,4,6-trinitrophenol, is also called picric acid.
|
Return the highlight to the selection mode icon |
C. Building a model of indigo
In the following steps, you use the Sketcher to build a model of indigo. In doing this, you review sketching while also learning some important new things:
| Create a new model space by choosing the "+" button on the Model Manager. |
| Rename this new model space "indigo". |
You're going to sketch the indigo model in this space.
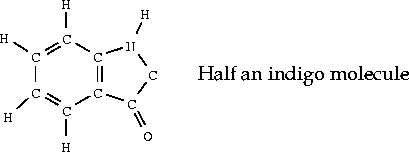
Hint: you may want to begin by using the template tool with the phenyl fragment.
|
Choose the Select All item from the Edit pulldown menu or the |
| Choose the Duplicate item from the Edit pulldown menu. |
A second copy of the molecule is pasted into the model window.
Only the selected molecule moves.
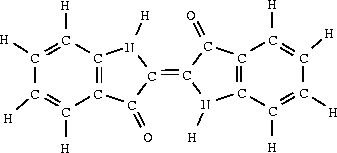
| Return the Sketcher to selection mode, and then close the Sketcher control panel. |
D. Measuring the indigo model
In the following steps, the Measurements control panel is used to measure the geometry of the indigo model from C. Building a model of indigo.
1. Measure the distance between two atoms.
| Open the Measurements control panel from the Geometry pulldown menu. Click on the Distance icon. |
As in the Sketcher, these icons determine the function of the mouse in the model window.
| Pick any two atoms. |
The distance between them will be marked on the model. You may need to rotate the model or zoom in on it to see the measurement clearly.
| Pick three atoms to measure angles, four to measure torsions or inversions, and six to measure the angle between two planes. |
| Choose the List Measurements button. |
Pick three atoms to measure angles, four to measure torsions or inversions, and six to measure the angle between two planes.
| Load the model Cerius2-Models/molecular-crystals/pigments/indigo_1.msi. |
This is a crystal structure of indigo.
| Perform the same measurements on this model as you made on the indigo that you sketched. Compare the results. |
| Finish by choosing the Delete Measurements button. |
This removes all the measure lines and values from the models.
E. Generating hard copy
In the following steps, you output the contents of the model window to a PostScript file, and then print a view of the trinitrophenol model in ball display.
| This exercise assumes that you are able to print grayscale PostScript files from your workstation. |
| Make trinitrophenol the current model and the only model visible. Set the display style to Ball. |
The model window background turns to white. (A white background will save toner when the image is printed.)
| If you want your name or any other text to appear on the image, type into the User Name and Title Text boxes. |
A message is written to the text window:
Starting to output image ...The PostScript image will be written to a file called -rinitrophenol.ps.
..................Image Captured
| If you are running Cerius2 on an SGI, choose the Preview button. (If you are running on another platform skip to step 4, below.) |
| Close the xpsview window by choosing Exit from the xpsview File menu in the top left corner. |
4. Select one of two methods to print the file.
There are two ways that you can print the file:
| On the Page Setup control panel, set the background Color popup back to Black. |
| It is also possible to use the SGI snapshot utility or the xv program to capture rgb images from Cerius2. |
5. Exit or continue to another tutorial.
Re-initialization resets all the Cerius2 control panels to their original defaults.Alternatively, if you want to stop now:
| Choose Exit from the File pulldown menu to quit from Cerius2, and choose the Exit button on the Exit Option... dialog box. |
Review
In this tutorial you learned how to:
Related material
The model manager is discussed in detail in the Cerius2 Modeling Environment. If you haven't already done so, you may want to read the introductory tutorial in Chapter 2 of that book. Other sections of that book that relate to the activities in this lesson include:
This chapter is divided into four parts:
In this tutorial, you learn about the Cerius2 Graphs card. Nearly all the Cerius2 materials science modules produce some output to the graph window. It's important that you understand how the Graphs card works so that you can get the most from your graphical results.
You can use the functions on the Graphs card:
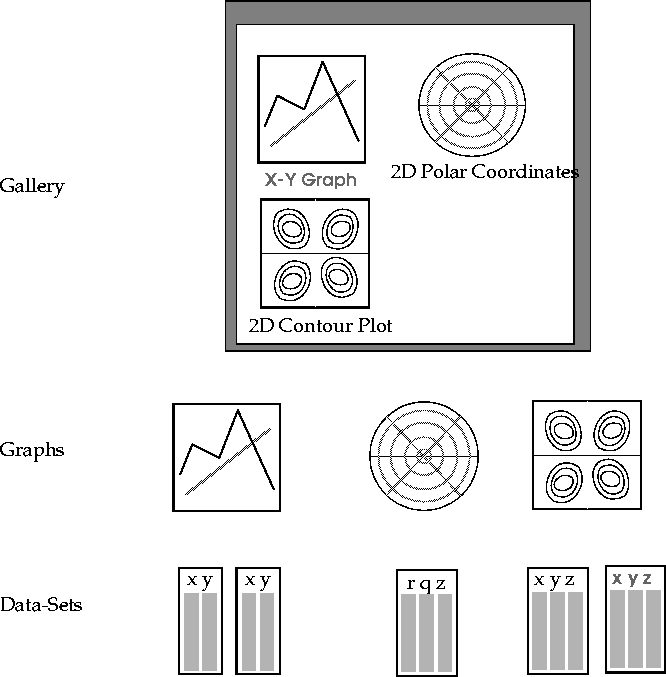
These are defined below and shown symbolically in the figure.
Data-set
At the bottom of the hierarchy is the data-set. The data-set must be one of three types:
At the middle level of the hierarchy is the graph. A graph is made up of one or more data-sets displayed on axes.
At the top of the hierarchy is the gallery. The gallery is the name given to the group of graphs on display in the graph window at a given time.
Summary of tutorial
The tutorial is divided into six exercises:
Files generated using Insight II can also be loaded; these have the extension .tbl and are described at the MSI website, www.msi.com.
The sample graphs that you load for this tutorial were created by the Blends module and the Sorption module. The contents in themselves are irrelevant for this tutorial, and you needn't have licensed the Blends or the Sorption modules to load or manipulate these graphs.
1. Load the graph files.
| Find the Graphs card (on the Tables & Graphs card deck). |
If the card is not already at the front of the deck, click on the card title to bring it to the front.
|
Open the Load Graphs control panel by choosing the File -> Load Graphs item on the Graphs card. Find the directory Cerius2-Resources /EXAMPLES/data. |
Be sure that the filename text box at the bottom of the browser box reads *.grf.
| Load the file EM_MM_DG.grf: pick its name on the browser box and then click the Load button. |
Loading Grapher file: ./Cerius2-Resources/EXAMPLES/data/EM_MM_DG.grf
Loading Done.
| Now load two more graph files from the EXAMPLES/data directory: EM_MM_Emix.grf and Loading.grf. Close the Load Graphs control panel. |
2. Arrange the graphs in the Gallery.
| Click on the Display check boxes next to Emix Fit v1 and Thermodynamics v1. |
Three graphs should now be on display in the graph window.
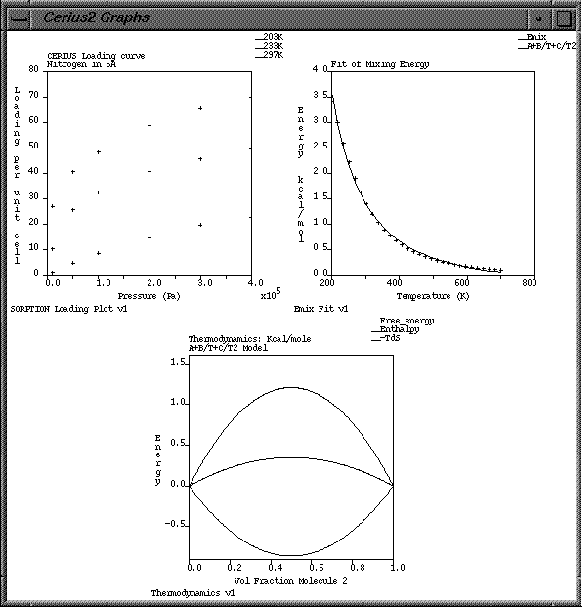
| Change the Gallery Width to 3 and the Gallery Height to 1, and note the result. Try some other width/height combinations before returning both settings to Auto. |
B. Using the Graph Manager and setting plotting attributes
Now that you've looked at the Gallery Manager and used it to edit the layout of graphs in the graph window, you learn how to edit the graphs themselves using the Graph Manager.
The top portion of your Data-Set Chooser should look like this:

| Click the Plotting Attributes... button at the base of the Graph Manager control panel to open the Plotting Attributes control panel. Change the Style popup (currently reading Point) to Line. |
Observe that the data plot style changes from points to a line.
| Use the Color popup (currently reading YEL) to change the line color to light green (LGR). |
| Edit the Label text box. |
Observe that the text for the green line is modified.
| Edit the Y Offset text box: for example, change it to 30. |
Observe the Y shift in the data plotted.
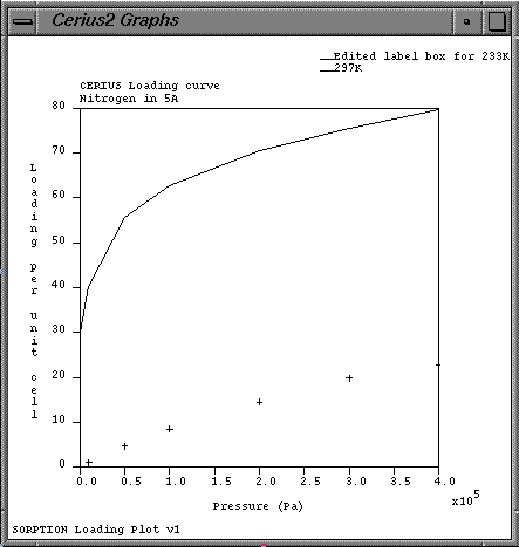
| On the Graph Manager control panel, select all the Sorption data-sets for both Display and Edit. |
| Return to the Plotting Attributes control panel. |
| Set the Y Offset to 0 (zero). |
| Experiment with the Style popup: set it to Histogram, to Delta, to Point, and finally to Line. |
| Close the Plotting Attributes control panel. |
C. Scaling graphs
In the following steps, you learn four ways to alter the scale of a graph:
| Open the Graph Scaling control panel by choosing the Graph -> Scaling item on the Graphs card or by choosing the Graph Scaling... button at the foot of the Graph Manager. |
|
Enter a value of -1e5 in the Min X text box and a value of -20 in the Min Y text box. |
Notice the effect on the graph axes.
![]() increases the zoom level
increases the zoom level
![]() decreases the zoom level
decreases the zoom level
![]() resets the graph to its original scale
resets the graph to its original scale
| Alter its value from 5 to 20% and notice the effect on the zoom rate. |
| Use the zoom buttons to adjust the graph scale so that the origin of the graph is not visible. Now use the scroll buttons until you can see the origin back on the graph. |
The scroll buttons work very similarly to the zoom ones. The arrows indicate the scroll directions, and the central button resets the scale. The scroll % text box is used to set the percentage of the axis scrolled in one button click.
| Try mouse scrolling: put the cursor within the axes of the graph, and move the mouse while holding down the middle mouse button. |
By moving the mouse diagonally, you can scroll in both the x and y directions at once.
| Try mouse zooming: put the cursor within the axes of the graph, and move the mouse while holding down both the <Shift> key and the middle mouse button. |
| Click the Zoom Reset button to return the graph to its original scale. |
| Click the Axis Scaling popup to view the choice of scale settings. Then click again using the right mouse button to display the help text that describes the five sorts of scaling. |
When you have finished reading the help text, click it to make it disappear.
| Try the Auto-XY setting, and then the Nice-XY setting. |
Notice the change in the Max Y value when you do this.
| Exit from the Graph Scaling control panel. |
D. Annotating graphs
In the following steps, you learn how to use the Annotation Option popups to display (or not display) graph axes and various text items. You also edit the text for the graph title and axes.
1. Experiment with the Graph Annotations.
| Open the Graph Annotation control panel by choosing Graph -> Annotation from the Graphs card or by clicking the Graph Annotation... button at the foot of the Graph Manager. |
| Change the popup options from Yes to No and back again so you can identify the various components of the graph: Axes, Label, Title, and Key. |
The Cell option only applies to 3D graphs.
| You will never want to select --> because it has no effect. However, Cerius2 may set a popup to --> to indicate that this option is not unique for the graphs in the edit list. |
| Edit the X Axis Label, the Y Axis Label, and Graph Title text boxes. |
Notice that the text on the graph is updated with the new labels and title.
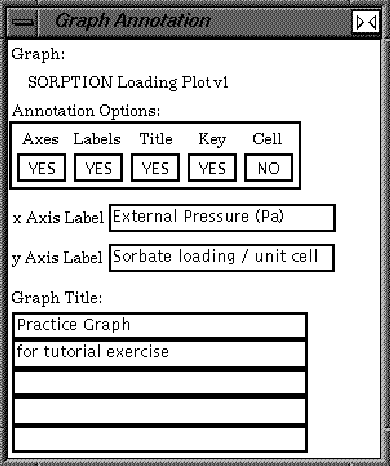
| Close the Graph Annotation control panel. |
E. Saving graph files
A graph (.grf) file can contain a single graph or multiple graphs. Each of the graph files that you loaded in section A (page 96) contained only one graph. In the following steps, you save a gallery of two graphs into one .grf file. Then, you reset the Graphs card and load back in the two-graph file that you saved.
1. Save multiple graphs into one file.
| On the Gallery Manager control panel select the Emix Fit and the Thermodynamics graphs to be displayed (and do not display the Sorption Loading Plot graph). |
| Open the Save Graphs control panel by choosing File -> Save Graphs from the Graphs card. Type two_graphs.grf into the filename text box and click the Save button. |
A confirmation message like the one shown below is written to the text window.
Saving Gallery File: ./two_graphs.grf2. Delete graphs from the Gallery.
Saved GRAPHS file.
| Choose Reset from the Graphs card, and choose Re-initialize Graphs from the dialog box to confirm the reset. |
Reset deletes all graphs from the Gallery Manager and returns all Graphs controls to their default settings.
| Open the Load Graphs control panel by choosing File -> Load Graphs from the Graphs card. |
| Load back the two_graphs.grf file. |
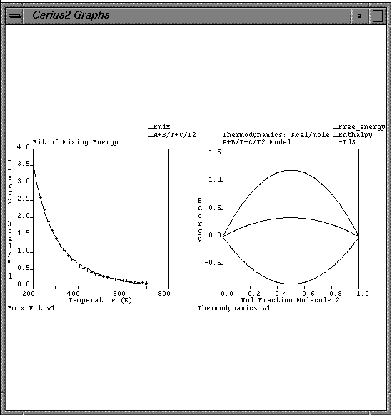
F. Creating new graphs and using expert functions
In this final section of the Graphs tutorial, you create a new User Graph and copy datasets from other graphs onto it. You also learn how to use the Expert Functions: Clean, Data Placement, and Keep Same.
In part E (page 108), you deleted all the graphs from the Gallery Manager by resetting the Graphs card. In the following steps, you learn how to selectively remove graphs from the gallery.
There is another version of the Emix Fit graph saved in the file Cerius2-Resources/EXAMPLES/data/EM_PM_Emix.grf. Here, you load this graph to illustrate the usefulness of the Keep Same and Data Placement options of the Graphs Expert Functions control panel. In this exercise, you load the second version from file, but, in practice, it will usually be a Cerius2 module that creates a new version of a graph and pipes it into Graphs. However, the principle is the same regardless of whether the graph is loaded or piped. The Data Placement control lets you decide how to treat new versions of the same graph. The Keep Same controls let you apply the graph formatting that you've specified for one graph to subsequent versions of the graph.
| Open the Gallery Manager control panel by choosing Gallery -> Manager from the Graphs card. Create a new User Graph by checking the third (empty) Display box in the Graph Chooser list box. |
This causes the list box to update with the name User Graph v1, and an empty set of axes appears in the graph window.
These two datasets are plotted on the User Graph.
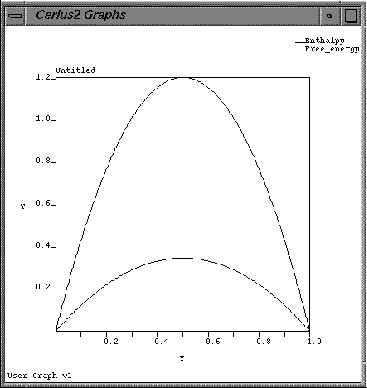
| Click the CLEAR button on the Gallery Manager/Graphs Expert Functions control panel. |
Clicking the CLEAR button deletes from the Gallery Manager all graphs that are not currently on display in the graph window. The Graph Chooser list box should now look like the figure below (the Thermodynamics and the User Graphs graphs are gone).
There are now two Emix Fit graphs listed in the Graph Chooser list box, and two Emix Fit graphs on display in the graph window.
| On the Gallery Manager control panel, set the second version of the Emix Fit graph to off. Then choose the CLEAR button of the Graphs Expert Functions control panel. |
This deletes the second version of the Emix Fit graph.
| Set the Data Placement option to Combine With Existing. Re-load the Cerius2-Resources/ EXAMPLES/data/EM_PM_Emix.grf graph file. |
| Now set the Data Placement popup to Replace Existing and reload the Cerius2-Resources/ EXAMPLES/data/EM_PM_Emix.grf. |
Now, the older version of the graph is overwritten by the new one. Replace Existing is the default for Data Placement; when a computational module creates versions of the same graph, the Replace Existing option prevents these many obsolete versions from accumulating in the gallery.
| Open the Graph Scaling control panel (Graph -> Scaling from Graphs card), and set the scale of the X axis to 300 - 600 and the scale of the Y axis to -1.3 - 2.0. |
| Open the Plotting Attributes control panel (Data -> Plotting Attributes from Graphs card), and set the Color popup to your favorite color. |
|
Set the following option values in the Graphs Expert Functions control panel:
Data Placement Additional Graph Keep Same: Graph Axes On Keep Same: Plot Attributes On
|
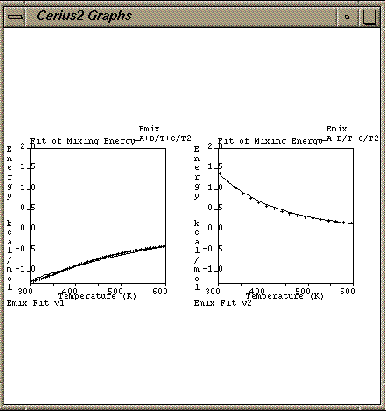
| Reload the file Cerius2-Resources/EXAMPLES/ data/EM_MM_Emix.grf. |
Because the Keep Same options are toggled on, the scale and plot attributes for the new graph are not the ones stored in the file, but instead, they are the same as the previous version of the graph.
| Re-initialize the program by selecting New Session from the File pulldown menu and choosing the Confirm button on the Re-initialize dialog box. |
| Choose Exit from the File pulldown menu to quit from Cerius2 and then choose the Exit button on the Exit Option... dialog box. |
o Load and save graph files
o Display graphs using the Gallery Manager
o Display data-sets using the Graph Manager
o Alter the data plot style and color
o Specify a Y-offset value for data-sets
o Edit text for graph titles, axes, and keys
o Use the Expert Functions to selectively remove graphs and to control version overwrites
What next?
In this tutorial exercise, you have encountered only the xy linear graph. Many Cerius2 modules also create 2D cartesian and polar graphs. These graphs can be manipulated (loaded, saved, modified) in much the same way as the xy graphs. When a polar or cartesian contour graph is selected for editing, the appearance of the Graph Scaling and Plotting Attributes control panels are automatically altered to show the special controls you need to modify 2D plots. You may now want to run one of the Cerius2 example files that creates a 2D graph and practice scaling it and modifying its plot attributes. For more information about 2D plots, see the Graphs chapter of the Cerius2 Modeling Environment.
You may now want to proceed to the tutorials in Cerius2 Materials Sciences Tutorials or Cerius2 Life Sciences Tutorials, or you may prefer to explore other Cerius2 modules on your own with the aid of the Cerius2 user's reference books and the on-line help text.
Related material
1. The Graphs chapter of Cerius2 Modeling Environment.
2. The File Formats appendix of Cerius2 Modeling Environment.
This chapter shows how to use molecular mechanics techniques to simulate the behavior of a system on an atomistic scale. The example used is a simulation of beta-D-glucose in water.![]()
Basic Molecular Simulation: Minimization & Dynamics
A. Build a model of the glucose molecule
1. Construct a glucose model
| To start Cerius2, enter cerius2 in a UNIX window. After Cerius2 starts, open the 3D Sketcher from the Build pulldown and construct a model of glucose. |
See Using the 3D Sketcher, for guidance on using the 3D Sketcher.
| Click and hold the CLEAN button in the Sketcher control panel to clean the structure. |
B. Minimize the glucose energy
During a molecular mechanics simulation the movement of the atoms (the changing of bond lengths, angles, etc., and the interactions between non-bonded atoms and molecules) are controlled by a mathematical function usually referred to as the forcefield. The accuracy of a molecular mechanics simulation depends critically on the choice of forcefield. The PCFF forcefield has been shown to give accurate models of a wide range of organic molecules and polymers and is used in this tutorial. Information about other forcefields and theoretical background about atomistic-level simulation using molecular mechanics can be found in the Forcefield-based Simulations book.
| Open the OFF SETUP card deck and locate the OPEN FORCEFIELD card. Load the pcff_300_1.01 forcefield. |
You may need to scroll down through a number of other forcefields to locate pcff. Note that references are written to the textport for your information.
| Change the Atom Labeling popup on the tool bar from NO LABEL to FFTYPE, and then to CHARGES. |
Note that the oxygen atoms and carbon atoms in different bonding environments have been identified as different atom types. Once the atom types and bonding environment were identified, partial charges were automatically allocated to each atom.
| Select the MINIMIZER card from the OFF METHODS card deck (it is usually the first card in this deck). |
| Open the Energy Minimization menu by selecting Run from the Minimizer card. |
| Select the Preferences... button to see the convergence criteria used. |
| Select the Run button to minimize the energy. |
C. Add water to the system
1. Create water molecules with the Sketcher
This sketches five or six isolated oxygen atoms --shown by default as small red stars.
| Select the H ADJUST button on the Sketcher menu to automatically add hydrogens to satisfy the oxygen valences. |
This produces five or six water molecules.
D. Simulating the behavior of the glucose/water system
1. Identify the atom types
| Return to the OFF SETUP card deck and open the Forcefield Atom Typing menu (select Typing/Atoms on the OPEN FORCEFIELD card). Calculate the atom types using typing rules. |
This identifies the atom types in the water molecules and assigns appropriate partial charges.
| You can select CHARGES from the Atom labelling popup menu to view these charges. Change back to NO LABEL before continuing. |
| Select the MINIMIZER card from the OFF METHODS card deck. Open the Energy Minimization card by selecting Run from this card. Reduce the Maximum Number of Interactions to 200 and Minimize the Energy. |
The most significant interactions between the glucose molecule and the surrounding water will be hydrogen bonds. The PCFF forcefield does not contain an explicit hydrogen bond term, but correctly models hydrogen bonding as a result of electrostatic and van der Waals interactions between non-bonded atoms. You can use the Edit H-bonds command under the Build pulldown menu to view the predicted hydrogen bonding.
| Turn on the Enable Automated Recalculation so that the changing hydrogen bonding pattern will be displayed during the molecular dynamics simulation. |
3. Simulate the system dynamics
| Select the Dynamics Simulation card from the OFF METHODS card deck and open the Dynamics Simulation menu by selecting Run. |
This model is fairly small so the number of dynamics steps can be increased to 2000 without making the simulation too time consuming. Each step simulates 0.001 ps, so the entire run will simulate the behavior of the glucose/water system for 2 ps.
| Select the Trajectory... button at the bottom of the Dynamics Simulation menu, or select Trajectory -> from the Dynamics Simulation card to open the Dynamics Trajectory Output menu. |
| Return to the Dynamics Simulation menu and select the Run Dynamics button. |
As the simulation continues, the structure on the graphics screen is updated every 25 steps.Notice the changing H-bond pattern. The graphs show that the temperature of the simulation fluctuates around the requested 300K.
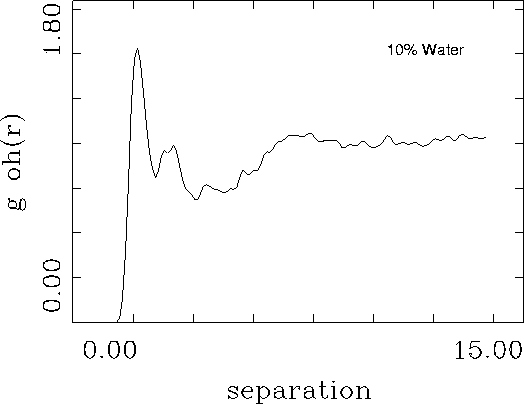
This simple simulation might be useful to provide insight into hydrogen bonding patterns and dynamic behavior. To obtain a more quantitative measure of hydrogen bonding between water and beta-B-glucose the calculation could be repeated with a larger number of water molecules using periodic boundary conditions. The resulting trajectory file could be analyzed to predict the pair correlation function between the hydroxyl groups on the glucose and the water.![]()
What to do next