Research Projects
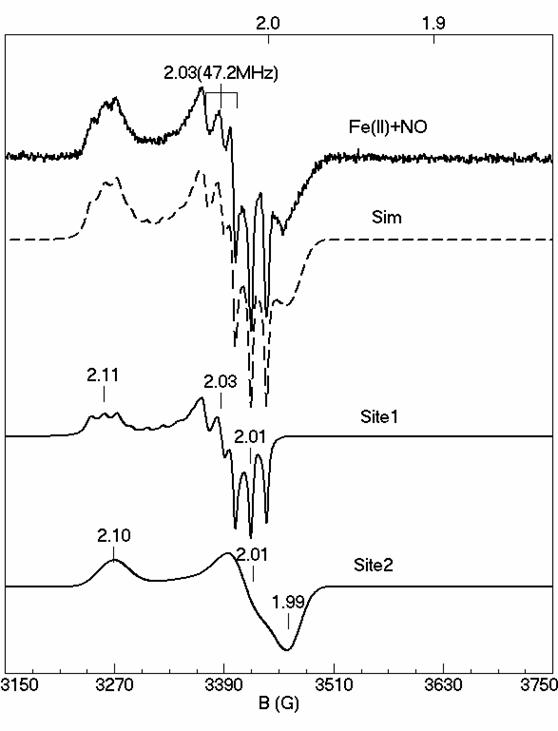
Tryptophan 2,3-dioxygenase (TDO)
Tryptophan 2,3-dioxygenase (TDO) is an enzyme responsible for dioxygenation of L-Tryptophan in human body. TDO and its isoenzyme IDO (indoleamine 2,3- dioxygenase) are the only two heme dioxygenases known in nature. TDO is a tetramer with a histidine ligated b-type heme in each of its monomer. Our recent studies have been towards understanding the structure-function relationship of the enzyme. Using EPR and Mössbauer spectroscopy, we have been able to characterize the ferric and ferrous substrate free and substrate bound states of the enzyme. In particularly, we have characterized the NO-adduct of the enzyme and using our computer simulation software, for the first time we have quantitatively identified the two overlapping species in the NO-adduct. Differences in the substrate free and substrate bound NO-adduct can give more insight into the structural changes that take place after substrate binding.
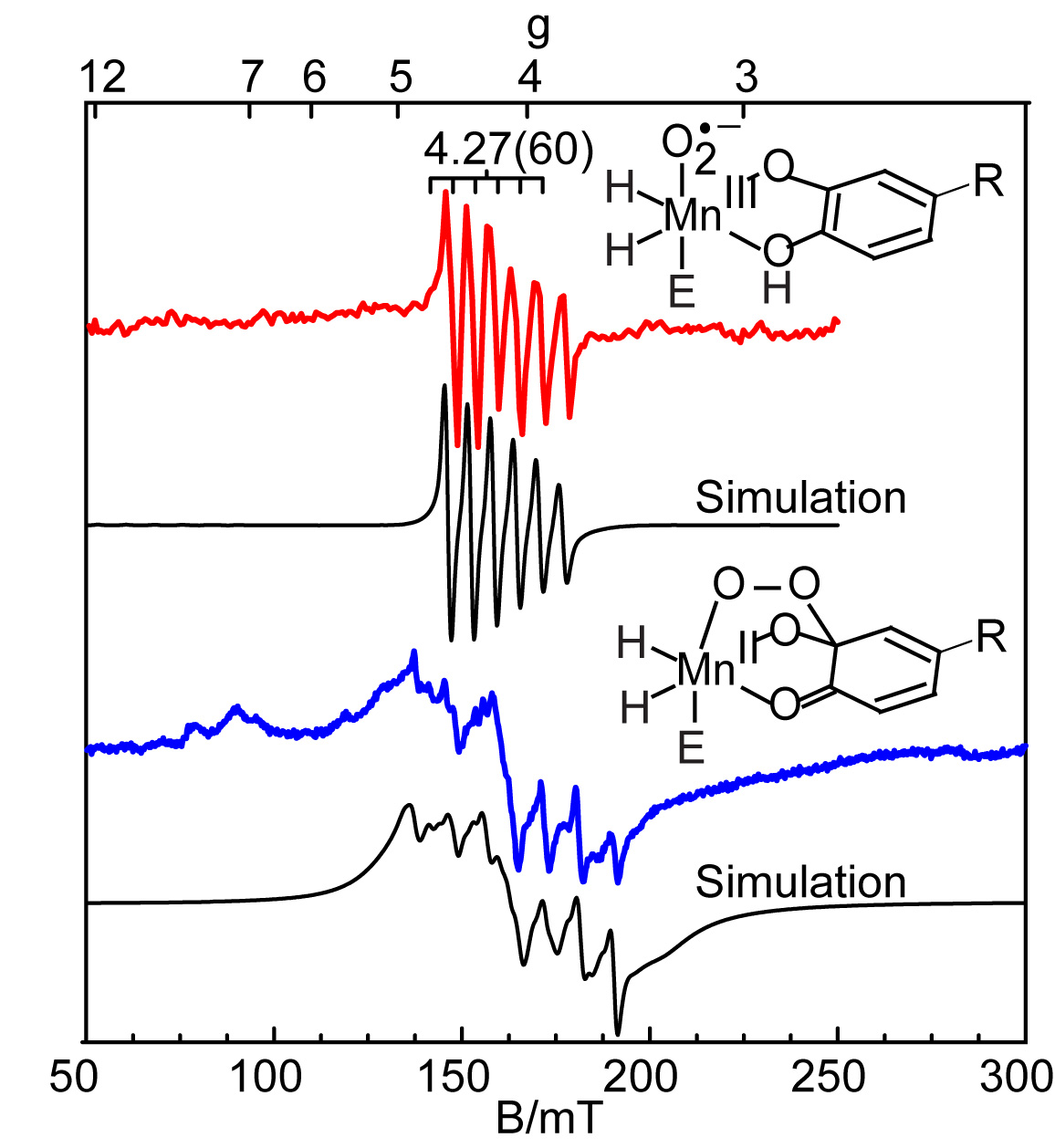
Manganese Homoprotocatechuate 2,3-dioxygenase (HPCD)
Extradiol catecholic dioxygenases catalyze the cleavage of the aromatic ring of the substrate with incorporation of both oxygen atoms from O2. These enzymes are important in nature for the recovery of large amounts of carbon from aromatic compounds. The catalytic site contains either Fe or Mn coordinated by a facial triad of two His and one Glu residues. Previous studies have shown that Fe(II) and Mn(II) can be interchanged in enzymes from different organisms to catalyze similar substrate reactions. Our studies focus on the Mn(II) substituted enzyme (MnHPCD). The combination of quantitative EPR spectroscopy and rapid-freeze quench experiments have allowed us to trap and identify four different Mn species within the catalytic cycle as the enzyme reacts with O2 and it natural substrate (homoprotocatechuic acid). Within the cycle we have identified two intermediates: a Mn(III)-radical species and a unique Mn(II) species, which we assign as Mn-alkylperoxo. Quantitative EPR simulations of these four Mn species allow us to assign kinetic rates to each step in the enzymatic cycle.
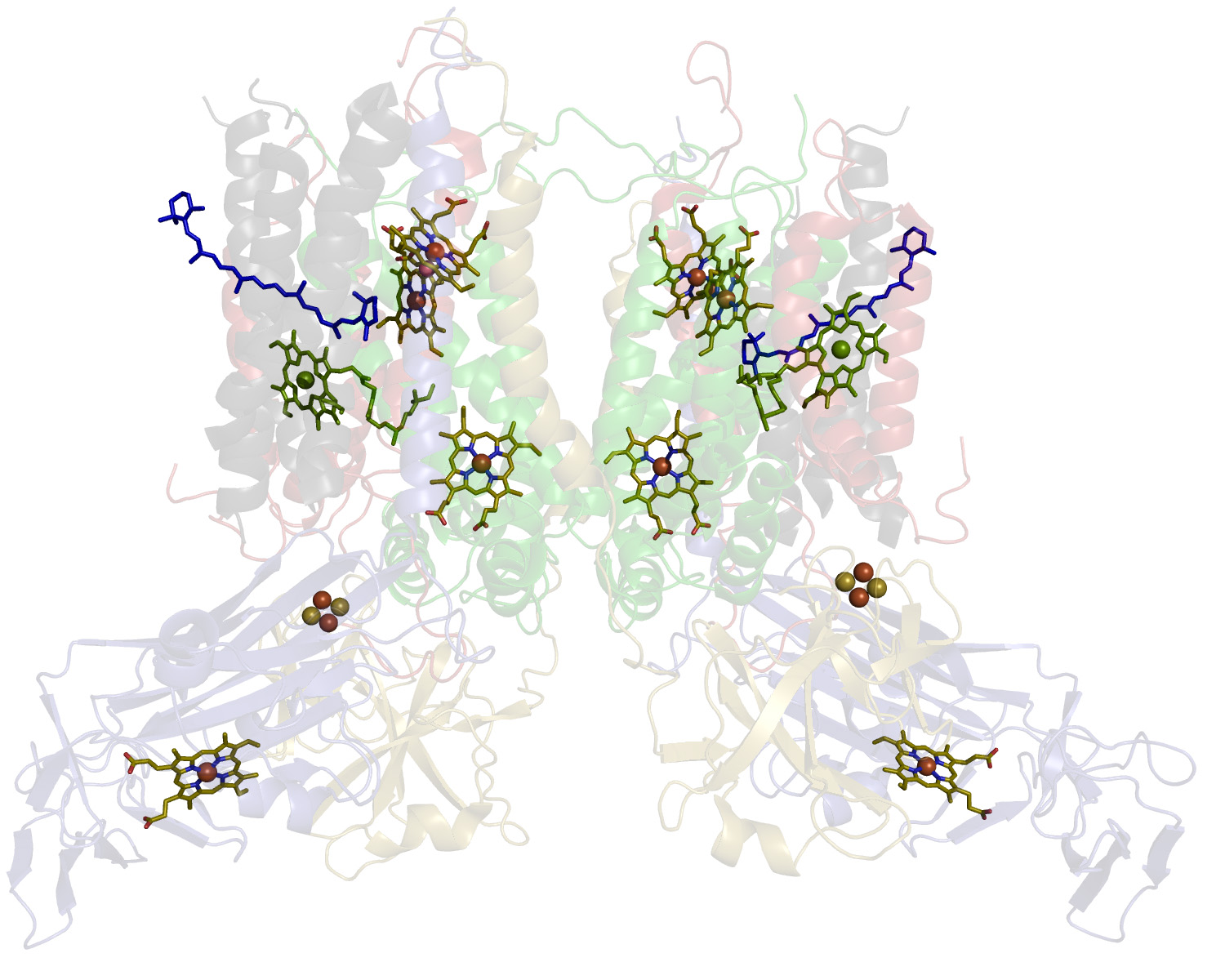
Cytochrome b6f complex
The 220 kDa dimeric cytochrome b6f complex of oxygenic photosynthesis provides the electronic connection between the two reaction centers, Photosystems I and II that are, respectively, coupled to NADP+ reduction and oxygen evolution. The electron transport functions of the b6f complex are coupled to proton transfer and generation of a trans-membrane proton electrochemical gradient, by mechanisms similar to those of the cytochrome bc1 complex of the respiratory chain and the photosynthetic bacteria, whose protein core is similar to that of the b6f complex. Prior to X-ray crystal structure analysis, each monomeric unit of the complex was known to contain six bound prosthetic groups, three hemes (f, two hemes b, bp and bn), one [2Fe-2S] cluster, and one molecule each of chlorophyll-a and carotene. Crystal structure analysis of the b6f complex from a green alga and a thermophilic cyanobacterium revealed the presence of an additional heme cn in the complex, which is covalently bound on the electrochemically negative (n)-side of the complex at a site very close to a b-heme (bn). Many EPR studies of the cytochrome b6f complex have been reported over past 25 years. In all these studies, signals with g= 4.3 were attributed either to an impurity species, or to a low-spin heme that lost an axial histidine, or to a mixture of isolated high-spin species. Using multifrequency, multimode EPR spectroscopy we demonstrated that all signals above g = 4.3 are in fact associated with an electronic interaction between a unique high-spin heme cn and a nearby low-spin heme bn. Frequency dependence of the signals above g = 4.3 and the observation of parallel-mode signals are both indicative of spin interactions in the complex. Heme cn has no axial protein ligands (unique in nature), thus we expected that small molecules such as cyanide would bind to it. Surprisingly, the addition of cyanide had only minor effects on the EPR spectra. In contrast, we have shown that a quinone analog NQNO binds at or near to heme cn. The two hemes remain spin coupled upon binding of NQNO, though the strength of the interaction decreases significantly. The electronic coupling implies that the heme cn/bn pair could function as a unit to facilitate 2-electron reduction of plastoquionone without generation of an energetically unfavorable semiquinone intermediate.
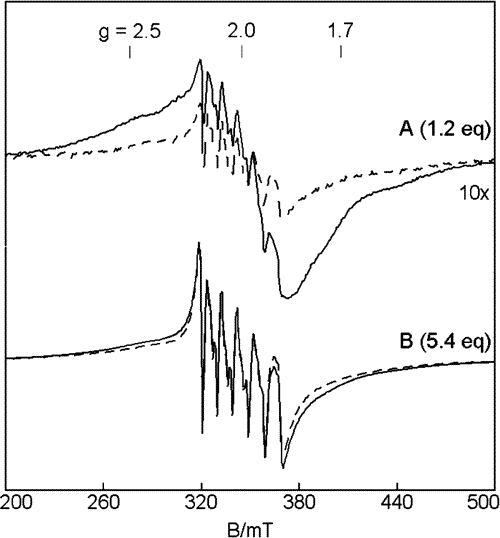
Manganese transport regulator (MntR)
The manganese transport regulator (MntR) is responsible for manganese homeostasis in Bacillus subtilis. The presence of Mn(II) inhibits the transcription of two Mn transporters MntH and MntABCD. Crystal structures show that MntR binds two manganese ions per homodimer, with Mn•••Mn distances ranging from 3.3 Å to 4.4 Å. Previous work on MntR had focused on the metal activation of MntR and showed tight DNA binding with an excess of Mn(II). In our lab we have focused on the binding affinity of Mn(II) to MntR to better understand the regulatory mechanism of the protein. Using EPR spectroscopy, the electronic structure of the active site can be directly probed. Mn(II) was found to bind to the protein in a cooperative binding mode with Kd = 160 μM. The exchange coupling is weaker than most characterized Mn2 sites with J = 0.2 cm-1. The weak coupling is consistent with the longer Mn•••Mn distance (4.4 Å) observed in the crystal structures. This work offers one of the first looks at how Mn concentrations are regulated inside the cell.
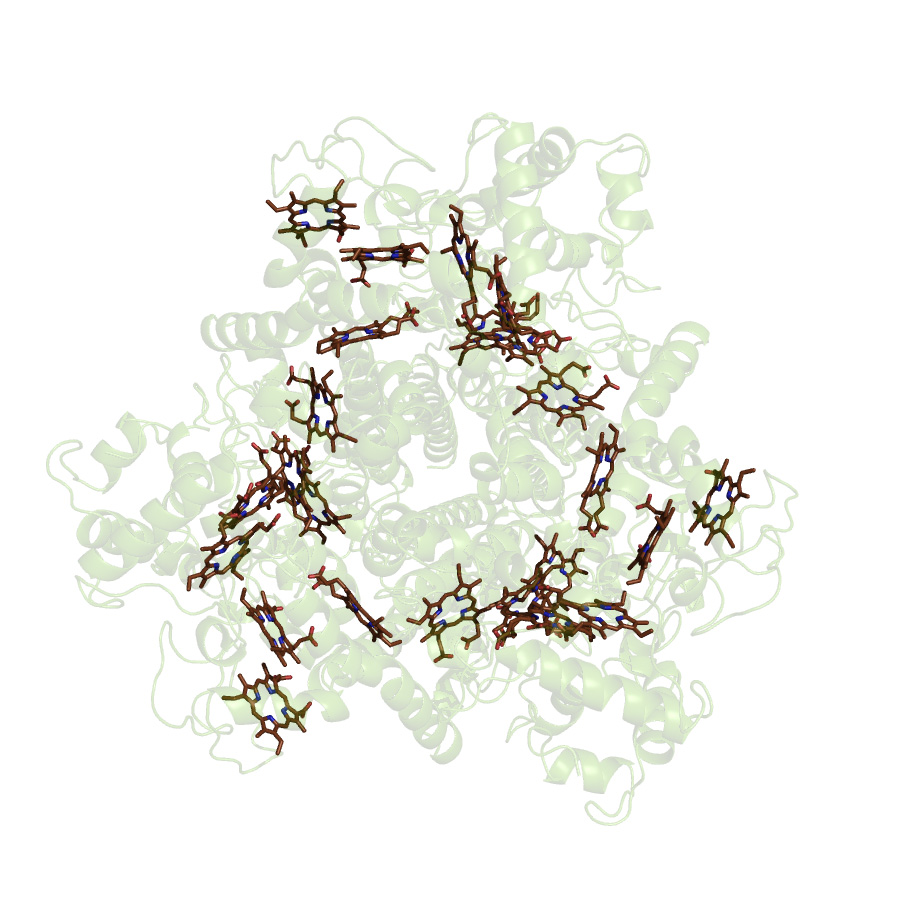
Multi-heme Proteins
The 24-heme enzyme hydroxylamine oxidoreductase (HAO) is the largest member of a new and growing class of multiheme proteins, which have conserved 3D structural arrangements of hemes despite absence of similarity in protein sequence or function. Some other proteins in this class include tetraheme cytochrome c554, fumarate reductase, pentaheme nitrite reductase, and octaheme tetrathionite reductase. The primary question that we are trying to address is: What is the function of so many conserved hemes in the mechanism of this wide range of enzymes? HAO catalyses a 4-electron oxidation of hydroxylamine, via a mechanism that is unique among all other known heme enzymes: during the turnover a net output of electrons is observed rather than a net input. Mechanistic studies of the substrate oxidation by HAO using rapid freeze quench EPR spectroscopy revealed that even a single turnover of the enzyme requires presence of an electron acceptor in the system despite availability of many Fe(III) hemes in HAO capable of taking electrons. This suggests that the function of the multiheme arrangement is an electron wire for efficient directed electron transfer rather than storage of electrons.
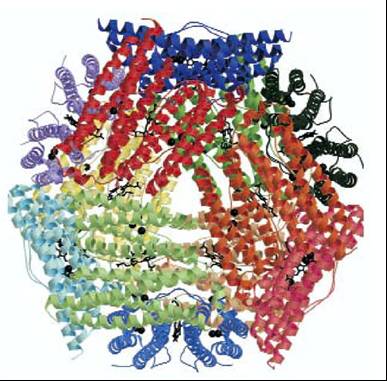
DNA binding protein from starved cells (DPS)
Besides catalases, prokaryotic cells use another class of proteins to remove
H2O2 and protect against oxidative stress. These proteins, termed DPS’s
have been shown to catalyze the mineralization of Fe2+ in the presence of
H2O2:
2Fe2+ + H2O2 + 2H2O --> 2Fe(O)OH(s) + 4H+
This reaction eliminates H2O2 and the two major reactants of Fenton chemistry
(H2O2 and Fe2+), which together can create dangerous hydroxide radicals.
In general, DPS proteins are dodecamers composed of twelve identical subunits
which form a hollow, protein cage. Each monomer has a di-iron center that
is similar to the ferroxidase center in eukaryotic ferritins. In ferritins,
this ferroxidase center is responsible for catalyzing the mineralization
of Fe2+ in the presence of O2 and is believed to play a major role in iron
storage. DPS proteins are believed to function primarily in preventing oxidative
stress. Recently, a DPS protein from Solfolobus solfataricus was characterized
and shown to form a hollow protein cage with tetrahedral symmetry and contain
a ferroxidase-like site. Though not previously known to bind manganese,
EPR of manganese-substituted DPS will prove to be a powerful spectroscopic
probe of the ferroxidase-like metal binding site. Additionally, structural
and functional similarities between the ferroxidase-like site in DPS and
that of manganese catalase indicate that it may function with manganese
bound in the active site. Current work in our lab has shown that DPS binds
manganese with a high affinity and we are currently exploring the potential
physiological relevance of the manganese-bound protein.
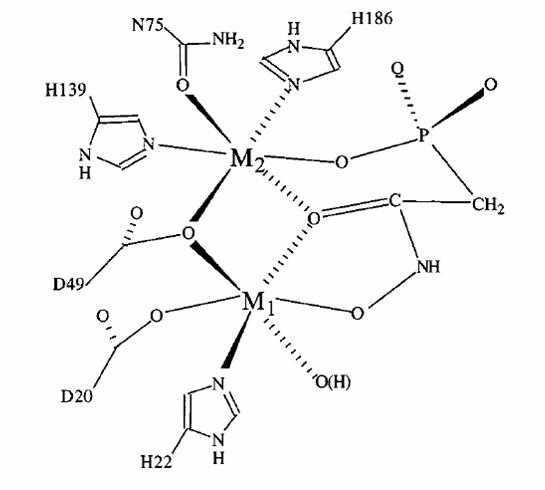
Lambda protein phosphatase (LPP)
LPP is a 25 kDa single polypeptide chain protein from the bacteriophage
lambda. It catalyzes the hydrolysis of a phosphoester bond from a serine/threonine
protein residue:
R-O-PO32- + H2O --> R-OH + HPO42- (R= Serine, Threonine)
This dephosphorylation reaction is important in the regulation of cellular
metabolism and general cell signaling. The proposed mechanism for this hydrolysis
involves nucleophilic attack of the phosphorous atom by a solvent molecule
coordinated to the dimanganese center. This coordination increases the Lewis
acidity of the water, thereby increasing its nucleophilicity. The nucleophilic
attack results in transfer of the phosphoryl group directly to the metal
center. This mechanism has been proposed based on kinetic and solvent isotope
effect data as well as support from a crystal structure of LPP with sulfate
bound in the active site. This structure was thought to represent an intermediate
formed during phosphoester bond hydrolysis, though spectroscopic evidence
of this is not yet available.
With the availability of a 2.1Å crystal structure, the ability to generate
large quantities of purified protein, and the capacity to generate site-directed
mutants, LPP is an ideal protein for the study of electronic structure/function
relationships in Serine/Threonine phosphatases. Though extensive EPR studies
have already been conducted on this protein, a thorough spectroscopic analysis
has yet to be done on the mononuclear and dinuclear manganese center. In
addition, spectroscopic evidence for the proposed mechanism has yet to be
presented. This work is currently underway in our lab.
Model Complexes
Our collaborators have developed an iron ligand system, [PhBPR3]Fe-Nx, designed to explore N2 fixation. This redox-rich iron system has been studied with EPR and Mössbauer spectroscopy in order to understand the electronic properties. Of particular interest are two terminal iron(IV) nitride species and a formally diiron(I) bridged, Fe(μ-N2)Fe species. The two iron species [PhBPiPr3]FeIV≡N and [PhBPCH2Cy3]FeIV≡N are diamagnetic and show identical Mössbauer spectra with an unusually large quadrapole splitting of 6.01 mm/s and a isomer shift of -0.34 mm/s. The diiron(I) complex shows a parallel mode EPR spectrum at g ~ 12, consistent with a S = 3 species, indicating a ferromagnetically coupled system. EPR and magnetization data suggests that the complex is a is a weakly coupled diiron(I) site. Understanding the electronic properties of these, and related complexes, allows further insight to the feasibility of N2 fixation by a single iron center.