| Quantum 1 Modules |
This chapter includes:
| For information about | See |
|---|---|
|
Configuring the DMol3 interface.
|
Quantum Chemistry Modules
|
|
Building or reading in models.
|
Cerius2 Modeling Environment and Cerius2 Builders.
|
After starting Cerius2, select the QUANTUM 1 card deck and then the DMOL3 card. This gives you access to functionality for setting up and running your DMol3 job and analyzing the results. The QUANTUM 1 card deck should now look something like:

This section explains how to use the Cerius2·DMol3 interface and does not discuss the DMol3 application in any detail. To learn more about DMol3, please refer to the documentation of standalone DMol3 in DMol3--Running in Standalone, DMol3--Keyword Descriptions, and DMol3--Keyword Reference. The most recent version of this documentation can be found at MSI's website:
DMol3 can be used to perform these basic tasks:
For all calculations, you of course first need to have a model present in Cerius2. For information on building and reading in models, please see Cerius2 Modeling Environment (published separately by MSI).
A typical DMol3 session involves several phases:
Who should read this section
If your model structure is satisfactory, you do not need to read this section and may proceed to Optional job control issues.
Read this section if you want information on:
The DMol3 program supports partial optimizations and transition-state searches. That is, the coordinates of some atoms are allowed to vary, while the positions of other atoms remain fixed during a calculation. This can decrease computation time considerably.
The DMol3 General Constraints control panel contains controls that mark selected atoms as fixed or free to move for geometry optimization. If the model is molecular, this control panel allows specification of fixed Cartesian coordinates and constrained distances, angles, and dihedrals. If the model is periodic, internal coordinate constraints such as distance may be set; however, bond angle and dihedral angle constraints for periodic models are not currently supported in the Cerius2·DMol3 module. For molecular dynamics and simulated annealing, only the Cartesian coordinate constraint is supported for both molecular and periodic models.
To modify a constraint (for example, change a coordinate constraint from fixed to varying or change the distance in a distance constraint from 1.28 to 1.05 Å), select the defined constraint in the list box, then (for example) change the Constraint Type popup and/or the Distance entry box and click the Modify Constraint action button. This updates the previously defined constraint.
Click the Show Constraint action button to highlight the atoms involved in the specified constraint.
Finding and setting the point-group symmetry
What symmetry is used for
With the DMol3 Symmetry control panel (which is accessed by selecting the Geometry/Symmetry menu item on the DMOL3 card), you can find the symmetry group of your model within a desired level of tolerance. You can also enforce exact symmetry upon your model and indicate to DMol3 that you want to use symmetry in the calculation.
If you also want to enforce exact symmetry by snapping the atoms to their exact symmetry positions and/or reorienting the model to the symmetry axes, make sure that the check boxes for Snap Atoms to Exact Symmetry and/or Re-Orient Atoms to Symmetry Axes are checked when you click the Find Symmetry action button.
If you find that you need to use very large tolerance values, you should probably use the Bond Geometry control panel (accessed by selecting the Move/Bond Geometry... item from the menu bar in the main Visualizer control panel) to manually adjust the geometry before using the DMol3 Symmetry control panel.
For some tasks, the symmetry found in this step may optionally be included in the DMol3 run.
Non-default job-control conditions are set with the DMol3 Job Control control panel, which you access by clicking the Job Control menu item on the DMOL3 card.![]()
Optional job control issues
By default, DMol3 jobs are run on the same machine on which you are running Cerius2. The job is run in the background and is not automatically monitored while it runs. If this is satisfactory, you do not need to read this section and may proceed to Setting up and starting calculations.
Read this section if you want information on:
Interactive
In interactive mode, Cerius2 displays the DMol3 output so you can monitor the progress of the jobs. However, you cannot do anything else in the Cerius2 interface until the job is complete, so this is useful only for jobs that you expect to finish quickly.
In background mode, the job runs without communicating with the Cerius2 interface, and you can quit Cerius2, allowing the job to run by itself. When you exit Cerius2, a status file is automatically saved so that, if Cerius2 is restarted, the job can be selected for monitoring or (if it has finished) the output files can be read in for analysis (see Analyzing DMol3 results).
In NQS mode, the job is submitted to the Network Queueing System, provided that this software has been installed, on the local or remote host. Once the job is queued, you can quit Cerius2, allowing the job to run by itself. An NQS-mode job can be selected for monitoring or file transfer in a later Cerius2 session.
Selecting a machine and base directory
By default, the DMol3 job runs on the machine on which you are running Cerius2. However, you may, for example, send it to a faster machine.
Click the Hosts arrow in the DMol3 Job Control control panel to obtain a pulldown listing machines at your site to which you can send the DMol3 job. The list contains all suitable hosts on your network (as defined in applcomm.db--for a description and example file entries, see the Cerius2 Installation and Administration Guide).
Depending on your site's setup, you might not have permission to run on all the machines listed in the pulldown, or you may need to specify a user ID and password to access some machines. Click the Options... pushbutton to access the DMol3 Job Control Options control panel, where you can input a User ID and Password before selecting your host machine.
Tip
|
If in doubt about any of the options relating to machines and network configuration at your site, please see your system administrator. |
You also need to specify a working (base) directory on the host machine if the default specification is not correct. For more information on how to do this, as well as whether files need to be transferred between machines in consequence, please see the on-screen help (click the right mouse button while the cursor is over the Base Directory entry box).
Setting up NQS-mode jobs
Setting Run Mode to NQS on the DMol3 Job Control control panel causes a More... pushbutton to appear. This button provides access to the NQS Control control panel, which allows you to set commonly used NQS flags (such as queue name and time and memory limits) and to set the names of the commands used to submit, monitor, and kill NQS jobs on the chosen host.
Monitoring and controlling running jobs
The Cerius2 DMol3 Job Status list box in the DMol3 Job Control control panel shows the filenames and directories associated with your DMol3 runs. For each job, the list shows the host name, datafile prefix, status (started, running, or complete for interactive and background jobs, or NQS_SUB, NQS_RUN, or complete for NQS jobs), process or NQS-request ID, and working directory. Click the UPDATE pushbutton to update this list. To remove an item from the list (if that job has completed), click the REMOVE pushbutton.
Tip
|
The status of the job is updated only if you click the UPDATE pushbutton, not if you merely close the DMol3 Job Control control panel or exit the Cerius2 session. |
Transferring files from one machine to another
If it is necessary to move output files from a remote machine to the run directory on your local machine (in most situations they are automatically returned at the end of a job), select the job in the Job Status list in the DMol3 Job Control control panel and click the TRANSFER pushbutton.
Note
|
The remote file system may actually be the same as your local file system (if, for example, it is NFS mounted the same way on both systems). In this case, no transfer of files is necessary. |
To start your DMol3 run from within the Cerius2 interface, access the DMol3 Run control panel by selecting the Run menu item on the DMOL3 card.![]()
Setting up and starting calculations
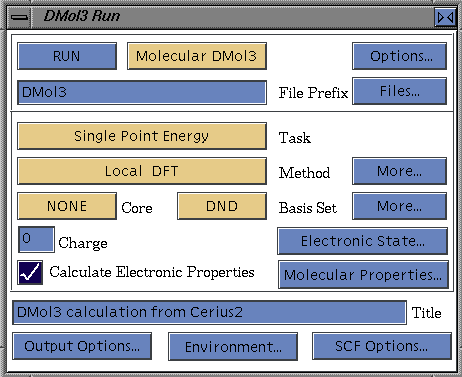
If the default options are satisfactory, you need only click the RUN pushbutton in the DMol3 Run control panel to start your run. Then proceed to Analyzing DMol3 results.
Read this section if you want information on:
For additional information on the options in all the control panels mentioned in this section, please see the on-screen help.
To change the default root name for files associated with your run, edit the contents of the File Prefix entry box in the DMol3 Run control panel. Alternatively, you can select a root name from existing datafiles using controls on the DMol3 Input File control panel (Saving, editing, and using input files). We refer to this root (or "seed") name as run_name in this documentation.
To change the default title for your run, edit the contents of the Title entry box in the DMol3 Run control panel. This descriptive text is included in the input and output files associated with your run, to aid in file identification.
For other aspects of file handling, access the DMol3 Input File control panel by clicking the Files... pushbutton in the DMol3 Run control panel.
The available tasks are:
A single-point energy task computes the SCF solution for the input geometry. By default, the least computationally demanding options are chosen, which sets up a calculation suitable for studying electronic structure (molecular orbitals, dipole moment, etc.).
A geometry optimization task optimizes your model to a stable geometry, that is, it locates a minimum in the potential energy surface.
If required, control over the optimization procedure is available on the DMol3 Geometry Optimization control panel, which is accessed by clicking the More... pushbutton next to the Task popup in the DMol3 Run control panel (when the Task is set to Geometry Optimization).
For information about constraining atoms during geometry optimization, please see Setting geometry constraints.
Frequency
DMol3 can evaluate frequencies by finite differences of analytic gradients. To perform a frequency calculation, set the Task popup in the DMol3 Run control panel to Frequency.
Control over the transition-state search (including the convergence criteria and the Hessian mode to follow) is available on the DMol3 Transition State Search control panel, which is opened by clicking the More... pushbutton next to the Task popup in the DMol3 Run control panel (when the Task is set to Transition State Search).
Geometry optimization and frequency
To perform a geometry optimization followed by a frequency evaluation, set the Task popup in the DMol3 Run control panel to Optimization & Frequency.
Transition-state search and frequency
To perform a transition-state search followed by a frequency evaluation, set the Task popup in the DMol3 Run control panel to TS Search & Frequency.
Tip
Gradient
To compute the energy and gradient for a given geometry (without optimization), followed by any requested properties, set the Task popup in the DMol3 Run control panel to Gradient.
Molecular dynamics
To perform molecular dynamics using the parameters specified in the DMol3 Molecular Dynamics panel, set the Task popup in the DMol3 Run control panel to Molecular Dynamics. Select the More pushbutton next to the Task popup to gain access to additional controls.
Simulated annealing
To perform various combinations of molecular dynamics protocols in series, set the Task popup in the DMol3 Run control panel to Simulated Annealing. You may perform melting, damping, quenching, constant energy and constant temperature molecular dynamics protocols. Select the More pushbutton next to the Task popup to gain access to additional controls.
Scan potential energy surface
To perform a scan of the potential energy surface, set the Task popup in the DMol3 Run control panel to Scan PES. This will allow you to vary one internal or Cartesian coordinate systematically, performing geometry optimizations at each value of the coordinate. Click the More button next to the Task popup to gain access to additional controls.
Calculation methods
The Method popup on the DMol3 Run control panel allows you to choose between several forms and parameterizations of the Hamiltonian. The default method is a local functional, PWC.
Atomic core representations
DMol3 supports several types of representations for the atomic core of atoms. Click the Core button to choose among the following options:
NONE
No core representation. Perform all-electron calculation.
ECP
Use relativistic Effective Core Potentials. This reduces the computational cost by collecting core electrons into a single analytical representation. Currently ECPs are available for elements from Z=21 (Sc) to 103 (Lr).
AE Relativity
Use all-electron with relativity. This does include relativistic effects that are important for heavy elements while retaining more accuracy than an ECP calculation.
Specifying a basis set
If you want to use a non-default basis set, select another one from the Basis Set popup in the DMol3 Run control panel.
For all choices, you may in addition click the associated More... pushbutton (next to the Basis Set popup in the DMol3 Run control panel) to further refine your basis set assignments, considering each element individually.
Specifying charge, spin, and occupations
Charge
Regardless of the type of calculation you want to perform, if your (non-periodic) model is not neutral you need to inform DMol3 of its charge by entering a value in the Charge entry box in the DMol3 Run control panel. Enter 0 for a neutral molecule, 1 for a singly charged cation, etc. For periodic models, the Charge must be zero.
Likewise, if you want to consider a particular spin multiplicity, click the Electronic State... pushbutton in the DMol3 Run control panel to open the DMol3 Electronic State Definition control panel. Use the Spin Polarization popup in this panel to specify a restricted or unrestricted spin calculation.
To set the initial spin densities on individual atoms, check the Initial Spin Populations check box in the DMol3 Electronic State Definition control panel and then click the Atomic Spin Densities... pushbutton that appears. This opens the DMol3 Starting Spin Densities control panel, which you can use to set and display the spin densities on selected atoms.
To specify occupations, use the Selecting popup in the DMol3 Electronic State Definition control panel. Other controls appear, depending on the setting of this popup, which allow you, for example, to read in fixed occupations from a file.
Calculating properties
There are two ways to specify the properties that DMol3 should calculate:
To specify properties while setting up the DMol3 calculation, make sure that the Calculate Electronic Properties check box in the DMol3 Run control panel is checked. If it is checked, then certain default properties such as Mulliken and Hirshfeld Analysis and HOMO and LUMO grids are calculated. You may also open either the DMol3 Molecular Properties or the DMol3 Periodic Properties control panel to specify more properties to calculate. These panels are specific to molecular or crystalline model types, respectively, and are opened by clicking the Molecular Properties... or Periodic Properties... pushbutton that appears in the DMol3 Run control panel when the Calculate Electronic Properties check box is checked. The properties specified in these panels are calculated as part of the DMol3 calculation.
If, after the DMol3 calculation is complete, you realize that you did not specify all the properties you wanted, you may specify and calculate them through the DMol3 analysis functionality:
SCF options
To change the default values for the parameters that control SCF convergence, click the SCF Options... pushbutton in the DMol3 Run control panel to open the DMol3 SCF Options control panel.
Molecular environment
DMol3 allows you to include the effect of an applied uniform electric field on molecular properties. To include such effects, use the DMol3 Environment control panel, which is accessed by clicking the Environment... pushbutton on the DMol3 Run control panel.
The solvation effects can be included by using the COSMO model. All the tasks specified for molecular calculations can be used with the COSMO model.
Specifying output and run options
Options to control the amount of output produced by DMol3 are found on the DMol3 Output Options control panel (accessed by clicking the Output Options... pushbutton in the DMol3 Run control panel).
Molecular dynamics options
The molecular dynamics options are defined in the DMol3 molecular Dynamics control panel. This panel is accessed by clicking the More... button next to the Task popup in the DMol3 Run control panel when the Task is set to Molecular Dynamics.
If the Use Default Time Step parameter is checked, the time step is automatically related to the smallest mass of any particle in the system propagated by molecular dynamics or simulated annealing. Otherwise, an explicit time step can be defined.
The Atom Mass parameter controls whether the same mass is to be used for all atoms. This option is useful in simulated annealing calculations to treat the update of all atomic positions on an equal basis. The PHYSICAL setting of the Atom Mass parameter allows you to employ atomic masses defined by the Cerius2 Visualizer (Use Cerius2 Atomic Masses). Otherwise, the internal defaults of the DMol3 application are employed.
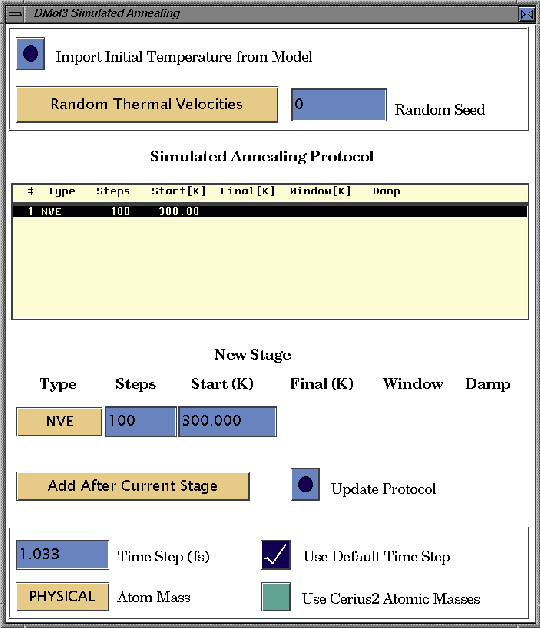
The top of the Simulated Annealing control panel deals with the initial atomic velocity assignment for molecular dynamics and simulated annealing calculations.
The middle of the Simulated Annealing control panel deals with the specification of the simulated annealing protocol. A simulated annealing protocol contains a set of stages which can be assembled in this control panel.
The bottom of the Simulated Annealing control panel defines the simulation time step and the atomic nuclear masses.
Every stage can be one of five distinct types:
Initial atomic velocity assignment
The generic velocity assignment control panel does not allow you to query the temperature that results from the combination of the three (i.e., thermal, linear and angular) contributions. This is possible in the Dmol3 Velocities control panel, opened by clicking the Query... button in the DMol3 Simulated Annealing panel, if Velocities from Model are supplied as the initial velocities. The DMol3 Velocities control panel further allows you to animate the assigned initial velocities. This functionality is accessible by clicking the Animation... button in the DMol3 Velocities control panel.
Simulated annealing protocol
The center of the DMol3 Simulated Annealing control panel sets the stages for the Simulated Annealing Protocol. The stages can be combined arbitrarily. With the exception of the NVT stage, the initial temperature of stages can be set to CONTINUE indicating that the current temperature of the simulation is used as initial temperature. For the first stage, this is only allowed if initial atomic velocities (and therefore an initial temperature) are specified.
If the Use Default Time Step parameter is checked, the time step is automatically related to the smallest mass of any particle in the system propagated by molecular dynamics or simulated annealing. Otherwise, an explicit time step can be defined.
The Atom Mass parameter controls whether the same mass is to be used for all atoms. This option is useful in simulated annealing calculations to treat the update of all atomic positions on an equal basis. The PHYSICAL setting of the Atom Mass parameter allows you to employ atomic masses defined by the Cerius2 Visualizer (Use Cerius2 Atomic Masses). Otherwise, the internal defaults of the DMol3 application are employed.
Scan potential energy surface options
The scan potential energy surface (PES) tool is used to vary one coordinate systematically, primarily for exploring reaction pathways. In cases in which the reaction coordinate is closely related to an internal coordinate, this is a simple but powerful way to scope a the pathway quickly; in cases of molecular adsorption, this is a convenient way to compute an energy profile as a molecule approaches a surface. The options are defined in the DMol3 Scan PES control panel. The panel is accessed by clicking the More... button next to the Task popup in the DMol3 Run control panel, with the Task set to Scan PES.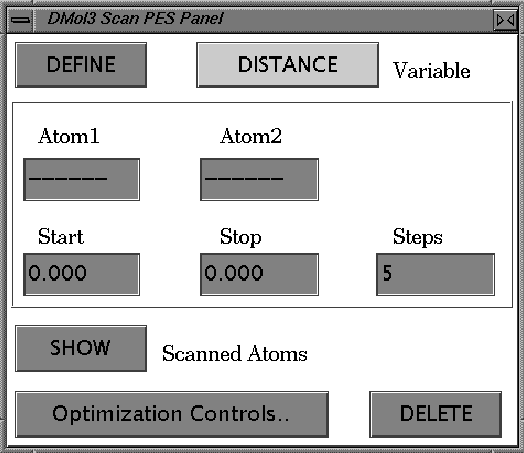
To specify the range of values to scan, use the type-in boxes labeled Start, Stop, and Steps. The coordinate will be varied systematically from the starting value to the stopping value in equally sized steps. The Start value is initialized to the current value of the coordinate; the Stop value is initialized to Start+0.2 for Cartesians or Start+10% for internal coordinates. (If Start+10% will exceed 180°, then the default will be Start-10%.) You can override the defaults by entering any real numbers for Start and Stop and any positive integer for Steps. Units are Ångstroms for distances and degrees for angles.
Clicking the SHOW Constrained Atom button will highlight the atoms involved in the coordinate and display the value of the internal coordinate. To delete the display of the coordinate value, click SHOW a second time.
To remove a constraint that you have defined, simply click the DELETE button.
To understand how to analyze the results of a Scan PES calculation, refer to the section Analyzing DMol3 results.
This section includes information on:![]()
Analyzing DMol3 results
Output from the last DMol3 job completed in interactive mode during this Cerius2 session is automatically selected for analysis. Any graphs produced (normal mode frequencies) are automatically displayed at the end of the run.
The list box in the DMol3 File Analysis control panel shows the DMol3 output files (run_name.outmol) in your current directory. You can browse other directories by using the popup above the list box.
By default, when you select a run_name.outmol file, the associated DMol3 model is automatically loaded into the model display window. If an infrared spectrum was produced, it is automatically displayed.
Information about the selected output file is displayed in the Summary of Calculation list box in the DMol3 File Analysis control panel after a file is selected.
The files that are generated by DMol3 in the Cerius2 interface (and required for data analysis) are:
The Display Selected Mode section of the DMol3 Vibrations control panel controls the Cerius2 model window. To animate a normal mode, select the ANIMATE tool. The Mode Scale entry box determines the maximum movement of any atom; the step number can be used to freeze the model at any step of the animation; and the total number of steps controls the smoothness and relative speed of the animation.
The Infrared Spectrum section of the DMol3 Vibrations control panel controls the Cerius2 graph window, which displays a plot of the infrared spectrum. The peak shape and width can be adjusted: select LORENTZIAN or DELTA to set the peak shape; enter a number in the Width entry box to adjust the Lorentzian peak width (in cm-1).
Finally, you may scale the calculated frequencies by a Frequency Scale Factor so that they agree better with experimental frequencies.
Analyzing orbitals and densities
Orbitals and densities are scalar functions that are defined in the continuous 3D space surrounding the model. In practice, they are evaluated on a fine grid of points that completely encompass the model. This produces a large amount of information, which is typically visualized as an isosurface; that is, the constant-value points surrounding the model are connected so as to form a surface. The data can also be visualized as slices showing the values of the function on a 2D plane cutting through the model.
Calculating orbitals
You can calculate (and display, see also Displaying orbitals and densities as surfaces) the molecular orbitals, appropriately oriented with respect to the displayed model.
To do this, you may specify the orbitals before starting the calculation (Calculating properties), in which case the grid files are generated as part of the original DMol3 calculation. Alternatively, after the calculation is complete, you may request that additional orbitals be generated.
Do this by selecting the Analyze/Properties/Molecular Properties menu item on the DMOL3 card to open the DMol3 Analyze Molecular Properties control panel.
You can control the resolution with which the grid is calculated, through a popup in the DMol3 Additional Molecular Orbitals control panel. LOW-resolution grids are faster to calculate and display, FINE- and X-FINE-resolution grids give higher- and highest-quality graphic output, and MEDIUM-resolution grids represent a compromise between these considerations.
Calculating densities
You can calculate (and display, see also Displaying orbitals and densities as surfaces) the electron density, appropriately oriented with respect to the displayed model. To do this, use the Electron Densities controls in the DMol3 Analyze Molecular Properties control panel, which is opened by selecting the Analyze/Properties/Molecular Properties menu item on the DMOL3 card.
You can calculate the total electron density, the deformation density, the density of alpha or beta spins, and/or the difference (i.e., spin density) between them. For a restricted open-shell DMol3 calculation, the alpha and beta spin densities are the same, so the beta density is not calculated.
Displaying orbitals and densities as surfaces
When you need to use this control panel
Finding your surfacing files
If you want to display a currently undisplayed surface, use the Files... pushbutton to access the DMol3 Surfacing Files control panel. Select the desired run_name_property.grd file and click the LOAD button. You can use the browser popup to access directories other than the current one.
Once you have a surfacing file loaded and if the default settings in the DMol3 Surfaces control panel are satisfactory, you can click the Create New Surface action button (in the DMol3 Surfaces control panel) to create and display that surface on your model. Make sure that none of the surfaces in the list box is selected if you want to create and display a new surface in addition to those already displayed.
An isosurface connects points in space that have the same value of some parameter. However, you can add an additional dimension to a surface, by making a property map, which displays the values of another property as different colors on an existing displayed surface.
Specify a property to be mapped by choosing the appropriate run_name_property.grd file from the list box and clicking the LOAD pushbutton. You can use the browser popup to access directories other than the current one.
If more than one surface is displayed or has been loaded, you may need to select the one on which to map the property, using the list box in the DMol3 Surfaces control panel (Displaying orbitals and densities as surfaces).
Click the Add Property action button in the DMol3 Property Maps control panel to display the property map.
Editing and displaying slices
To edit and display a 2D slice through the 3D grid of orbitals or density for your model, use the DMol3 Slices control panel, which is accessed by selecting the Analyze/Slices menu item on the DMOL3 card.
If necessary, choose the orbital or density file to be surfaced by clicking the Files... pushbutton to access the DMol3 Surfacing Files control panel (Finding your surfacing files). Select the appropriate run_name_property.grd file from the list box and click the LOAD pushbutton. You can use the browser popup to access directories other than the current one.
Slice specification and display
Once you have loaded a .grd file and if the default settings in the DMol3 Slices control panel are satisfactory, you can click the Create New Slice action button to create and display a selected slice for your model.
A slice is defined by its position and direction. The default position and direction are chosen so that the slice passes through the best-fit plane to the whole model or any selected atoms. A slice is created with a default position and direction and can be returned to the default position or direction by clicking the appropriate reset action button.
![]()
Clicking the More Editing Options... pushbutton gives you access to the DMol3 Slice Preferences control panel, which contains additional controls that affect slices.
To create a 2D contour graph corresponding to a selected slice plane, click the Create Slice Plot in Graph Window action button in the DMol3 Slices control panel.
Molecular dynamics and simulated annealing trajectories
Trajectories from molecular dynamics and simulated annealing runs are stored in .his and .arc files. You may use the Analysis tools in either the OFF Methods or the Discover module in Cerius2 to analyze the trajectory. First load the .his or .arc file -- just as you would when analyzing a classical molecular dynamics trajectory.
Note
|
The potential energy in the .his file is the DFT total energy of the model, the same as the total energy obtained from a DMol single point energy calculation on the same configuration. |
Scan Potential Energy Surface results
Results from a Scan PES calculation are stored in an archive file, run_name.arc. When you open the DMol3 run_name.outmol file for analysis, a graph of energy vs. geometry is automatically displayed. You can select any point on the graph to display the corresponding geometry.
This section contains tutorials, which illustrate the application of several features of DMol3 to typical modeling problems:![]()
Tutorials
Lesson 2: Exploring the CH4 + Cl -> CH3 + HCl reaction with DFT MD in DMol3
Lesson 3: Using COSMO-RS to compute thermodynamic properties of binary and ternary mixtures
In Lesson 1 COSMO and COSMO-RS are used to compute the solvation energy, vapor pressure and partition coefficients of methanol. In Lesson 2 the molecular dynamics capability of DMol3 is used to study a chemical reaction. In Lesson 3 COSMO-RS is used to compute thermodynamic properties of binary and ternary mixtures. These procedures can be used as templates and applied to more complicated systems.
Lesson 1: Computation of solvation
energy, vapor pressure and partition coefficients for Methanol using COSMO
and COSMO-RS
1. Sketch and minimize methanol
| Select the Build/3D Sketcher... command from the Visualizer menu bar to open the Sketcher control panel. Using the control panel, sketch CH3OH and clean the structure. |
| Go to OFF SETUP card and click on OPEN FORCE FIELD/Load.
This opens up the Load Force Field pulldown. Choose the
COMPASS forcefield and click LOAD. Close the Load Force
Field pulldown.
Go to OFF METHODS and click on MINIMIZER/Run. This opens up the Energy minimization panel. Click the Minimize the Energy button. This geometrically optimizes the methanol molecule. Once the molecular structure is optimized, close the Energy Minimization panel. |
Note: If you do not have the COMPASS
forcefield, you could follow the above step by using the pcff_300_1.01
or the cvff_300_1.01 forcefield.
2. Run the geometry optimization in gas phase using DMol3
Now that you have a good initial geometry from energy minimization with
the COMPASS forcefield, you are ready to further refine the structure
using the first principles Quantum mechanical methods of Dmol3.
Note: The above settings were chosen in anticipation
of running COSMO-RS later on in this tutorial. The COSMO-RS
parameters were developed using the same settings of DMol3 as described
above.
Monitor and analyze the results.
| Click on DMOL3/Job Control. From the list of jobs in
the DMol3 Job Control panel, choose ch3oh_g and click UPDATE.
This should tell you if the job is still running or is completed.
Once the job is completed you can analyze the results. Click on DMOL3/Analyze/Files. From the list of *.outmol files in the DMol3 File Analysis panel choose ch3oh_g.outmol and click SELECT. This should load the DMol3 optimized structure of methanol onto the model window, and at the same time give a brief summary of the calculation in the Summary of Calculation box. In particular note the total energy, which should be something like -115.757xxxx Hartrees. |
3. Run DMol3 on Methanol in the solvent environment of water.
Note: Normally you should perform only Single
Point Energy calculations with COSMO on, because in most cases the structural
change from gas phase geometry is insignificant. However, for polar
molecules, and in particular for Zwitterionic systems, the molecular geometry
may be strongly affected by the presence of the solvent. In such situations
it would be important to perform Geometry Optimization with COSMO turned
on. However, the gradients in the presence of a solvent are not as precise
as in the gas phase, and therefore you should use COARSE criteria when
performing geometry optimization calculations in the presence of a solvent.
| Monitor the job through the DMol3 Job Control panel. Once the run is
finished, SELECT the file ch3oh_water.outmol from
the DMol3 File Analysis panel. Note the total energy in the Summary
of Calculation box. It should be more negative than the total energy
in the gas phase, something like -115.765xxxx Hartrees.
Actually, calculations with COSMO produce four different Total Energies (due to various corrections). All of these can be found toward the end of the *.outmol file. For example, in the ch3oh_water.outmol file, these are printed as: 1. Total Energy (au) [TE]
2. Total Energy corrected (au) [TE(corr)]
3. Total energy (au) [TE]
4. Total energy corrected (au) [TE(corr)]
Expression in 1 is the total energy the system SCF converges to, and appears in the Summary of Calculations box. Energy in 2 accounts for a small correction due to the spilling of charge outside the COSMO cavity. Expressions 3 and 4 are the same as expressions 1 and 2 respectively with additional correction due to non-electrostatic solvation energy. Use Expression 2 to study reactions in solutions, and Expression 4 for comparing energies of conformers. |
Wait for the COSMO-RS job to finish.
| In a UNIX window, open the file ch3oh_cosmors.outmol using your favorite editor. |
The results should appear as follows at the
end of the file:
COSMO-RS results for compound:
ch3oh_cosmors
Temperature = 298.15 K
Delta G of hydration (in kcal/mol):
-4.60
log(vapor pressure) (in bar):
-0.61
Log K ( octanol / water ):
-0.76
Log K ( hexane / water ):
-2.76
Log K ( benzene / water ):
-1.91
Log K ( diethylether / water ):
-1.11
Lesson 2: Exploring the CH4 + Cl -> CH3 + HCl reaction with DFT MD in DMol3
1. Build the models
|
Now move the Cl atom to within 3 Å of one of the H atoms in CH4. (Use the control panel accessed with the Visualizer command Geometry/Measurements to measure the distance). |
This hydrogen atom will be referred to as "the H atom" in the following text.
|
Go to the QUANTUM1 card deck and then to the DMOL3 card. Select the RUN item to open the DMol3 Run control panel. |
|
Set the Task to Molecular Dynamics, the Method to Local DFT, and Basis Set to DNP. |
|
Click Electronic State... to bring up the DMol3 Electron State Definition control panel. Set Spin Polarization to UNRESTRICTED and Occupations to THERMAL. |
|
On the DMol3 Run control panel, uncheck the Calculate Electronic Properties option and change the seed filename under the RUN button to ch4_cl. |
|
Click the More... button adjacent to Task to bring up the DMol3 Geometry Optimization control panel. Set the first popup to CONSTANT NVE and change Random Thermal Velocities to Velocities from Model. |
|
Click Assign... to bring up the Assign Velocity control panel. Click the Assign Thermal Motion button to assign random velocity to all atoms in the system. |
|
Click the Show Velocity Vectors button at the bottom of the control panel to see the velocity assignment. |
|
Click the Set Linear and Angular Momenta to Zero button. |
|
Select the H atom and, holding down the <Shift> key, select the Cl atom. Then click Define Direction from Selected Atoms to assign the path of reaction for C and Cl atoms. |
|
Select the Cl atom. Enter -30 in the SPEED text box to assign a 30 Å/ps velocity in the opposite direction of the Cl atom. With the Cl atom highlighted, click the Assign Net Linear Velocity button. |
|
Click the More... button adjacent to Task to open the DMol3 Molecular Dynamics control panel. Set Number of MD Time Steps to 100 and Initial Temperature (K) to 2000. Close the control panel. |
|
Go back to the DMol3 Run control panel and click the RUN button to launch the job. |
3. Analyze the result during and after job is finished
|
Now go to the OFF METHODS card deck and select the ANALYSIS card. Click the Input item to open the Analysis Input control panel. |
|
On the ANALYSIS card, select the Analyze/Statistics item to bring up the Analysis Statistics control panel. In the Select Properties list box, select Total Energy and click the Profile button. |
|
Use the panel accessed by selecting the Geometry/Measurements... command in the Visualizer window to measure the C-H and H-Cl distances. |
|
Go back to the Analysis Statistics control panel. In the Select Properties list box, select Total Energy, hold the <Crtl> key and click Distance, then click the Profile button. |
|
On the ANALYSIS card, select the Show Frames item to open the Analysis Show Frames control panel. Click the right arrow to play the trajectory in the .arc file. |
Lesson 3: Using COSMO-RS to
compute thermodynamic properties of binary and ternary mixtures
The COSMO-RS procedure can be used to compute a number
of thermodynamic properties of mixtures including:
It is strongly recommended that you perform the tutorial "Computation of solvation energy, vapor pressure, and partition coefficients for Methanol using COSMO and COSMO-RS" before this one. The former tutorial has more detailed explanations of various steps and settings involved in a COSMO/COSMO-RS calculation, and includes information necessary for such calculations with other systems.
1. Prepare the COSMO data for ethanol
and benzene
| Select Build/3D Sketcher... from the Visualizer menu bar to open the Sketcher control panel. Using the control panel, sketch CH3CH2OH and clean the structure |
| Go to OFF SETUP card and click on OPEN FORCE FIELD/Load.
This opens up the Load Force Field pulldown. Choose the
COMPASS forcefield and click LOAD. Close the Load Force
Field pulldown.
Go to OFF METHODS and click on MINIMIZER/Run. This opens up the Energy minimization panel. Click the Minimize the Energy button. This geometrically optimizes the ethanol molecule. Once the molecular structure is optimized, close the Energy Minimization panel. Next optimize the structure in gas phase using DMol3. |
| Repeat the above steps for benzene. Use a File Prefix benzene_g for the gas phase calculation and benzene for the COSMO calculation. |
After the jobs have finished, continue with the next step.
2. Prepare the COSMO-RS input file
| Create a file called CRS_INPUT in the directory that you plan to use for running the standalone DMol3 job. |
BINARY ethanol and benzene at two temperatures VPant 0 2 1 ethanol.cosmo -155.0961210 8.1122 1592.864 226.184 benzene.cosmo -232.3147340 6.87987 1196.76 219.161 2 2 298.15 398.15 |
| Save the file CRS_INPUT in the directory that you plan to use for running the standalone DMol3 job. |
3. Prepare the input data for DMol3
| Using your favorite text editor, edit one of the DMol3 input files that was used for the molecules in the binary mixture, say benzene.input. |
| Save the input file, in this case, benzene.input. |
Be certain you have the CRS_INPUT file, the DMol3 input file, and the saved files from the two earlier runs all in the same directory. Then run DMol3 from this directory. So, from the UNIX prompt (denoted by "%" below), type:
% benzene.run &
The output file, benzene.outmol,
contains results for a number of properties including excess enthalpy and
excess free energy as a function of concentration, at each of the requested
temperatures.
A portion of the output from this calculation
appears below:
| =============================================================
Thermodynamics of Binary System =============================================================
Compound #1 : ethanol
Vapor pressure of pure substance (in bar): 1
0.05899
x, molar concentration
Results for Compound #1 : ethanol
Results for Compound #2 : benzene
Excess Enthalpy (ex_ent) and Free Energy (ex_fen)
|
Decide which of these properties you wish to plot. Using the text editor, cut out the data you wish to plot, and save it to a file. You can then use a number of different utilities such as xgraph on UNIX or Microsoft Excel® to plot the data.
6. Details of the CRS_INPUT file
A sample CRS_INPUT file for the ethanol/benzene
system was presented in step 2. In this
section we discuss the details of the input parameters in this file. The
basic form of the input is as follows. Keywords are given in the order
they appear in the CRS_INPUT file. This order must be maintained whenever
you create a new input file.
*The Antoine equation provides an approximate formula for the log of pressure as function of temperature:
where A,B, and C are empirical parameters, and C is the temperature in degrees Celsius. Several sources for these parameters are available on the world-wide web including
http://www.glue.umd.edu/~nsw/ench250/antoine.dat
http://chemengineer.miningco.com/library/weekly/aa071497.htm
To rerun the above example without using the Antoine
coefficients, the input would be:
| BINARY ethanol and benzene at two temperatures
VPexp 0 2 1 ethanol.cosmo -155.0736739 benzene.cosmo -232.2865776 2 2 298.15 eth1 benz1 398.15 eth2 benz2
|
where eth1 and benz1 are the observed pressures at 298.15ºK of
ethanol and benzene respectively; and eth2 and benz2 are the pressures
at 398.15ºK.