
| Cerius2 Modeling Environment |
These general-purpose basic and advanced building and editing tools are directly available from the Build, Edit, and Move pulldown menus on the main control panel's menu bar.
Why read this section
![]()
Basic building
Related information
Basic editing tasks, including changing element types, bonding, and model conformation, are presented under Basic editing. The final task you would perform for building almost any model would be to optimize its configuration (Refining the conformation). Storing models in files is presented under Saving model structure files.
Selecting atoms is presented under Selecting atoms and groups of atoms.
Tasks that may be less often or less typically performed, such as editing atom attributes and changing defaults and preferences, are covered in Advanced building and editing.
If you have never used Cerius2 before or are unfamiliar with the terminology used to indicate how to use its interface, please go through the model-building example in Introducing Cerius2 before continuing with this section. How to start Cerius2, the names of parts of its interface, as well as basic mouse usage and how to access on-screen help, are covered in The Cerius2 Interface,
Starting to construct a new model
Strategy
Why develop a strategy
You can probably construct a model of almost any small model you want, simply by straightforward trial-and-error use of the 3D-Sketcher. This procedure will probably be good enough if you have to build only a few models. However, Cerius2 building tools typically offer several ways of accomplishing the same end.
| This guide does not necessarily replicate control-specific information that can be found by using the on-screen help. |
A few of many possible approaches include:
How it works
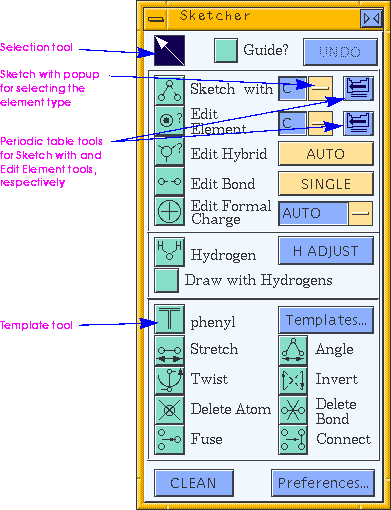
Clicking any of the icon tools on the Sketcher control panel alters the function of the left mouse button. Selection mode, when the selection tool (the arrow in the upper left of the Sketcher control panel) is highlighted, is the default for cursor behavior. Clicking other icon-type tools enables you to use the cursor, for example, to place atoms, manipulate angles, or change an atom's element type.
In some Sketcher modes, a property of the structure can be changed by moving the mouse in the model window. Generally, dragging left-to-right or bottom-to-top while holding down the left mouse button increases a value (e.g., a bond length), and dragging in the opposite direction decreases the value.
An UNDO button, provided at the top of the control panel, can be used to cancel one or a series of previous commands for many types of operations. The UNDO button is greyed out when it is not usable.
Checking the Guide? check box gives you on-screen help (in the upper left corner of the model window) on the 3D-Sketcher.
Detailed examples of using the Sketcher control panel to build caffeine (Building a model--caffeine) and ferrocene (Building another model--ferrocene) are included in this guide.
Other basic building tools
Other building tools are also accessed from the Visualizer's Build menu. Use of some of these is covered under Advanced building and editing, but others have functions that are similar to those in the 3D-Sketcher control panel. Among the latter are the Add Atom..., Edit Atoms..., Edit Bonds..., and Edit H-Bonds... menu items.
Most of these enable better control over exactly how the controls operate than does the 3D-Sketcher control panel and may therefore enable faster building under certain circumstances.
Adding atoms and bonds
The simplest procedure for building the basic structure of a model is to draw in the model window, manually placing atoms (connected by single bonds) at desired positions (see Adding atoms and single bonds). The model is then generally edited, and finally its conformation is optimized (Refining the conformation). Precise positioning of atoms as they are added is also possible (Precise positioning of atoms during building).
| This section refers to using the Sketcher control panel (The 3D-Sketcher), unless otherwise indicated. |
Selecting the element type
The sketching element type is listed in a text entry box to the right of the Sketch with tool:![]()
To begin sketching, simply click the Sketch with tool to activate it (if it is not already active--when an icon-type tool is active, it is colored dark purple.) When you move the cursor into the model window, it is shaped like a triangle, indicating that you are in sketch mode, that is, that clicking the left mouse button places an atom and/or bond rather than selecting an atom (or some other activity). The element that is used in sketching is specified as described under Selecting the element type.
You can use the Sketch with tool to start building a new model and to add atoms to existing models.
| When you click an atom, Cerius2 beeps to indicate that you succeeded. |
| You do not need to specifically add hydrogen atoms--this can be done automatically while sketching or (more quickly) after sketching is completed (Adding hydrogens). |
Other sketch-mode functionality
While in sketch mode, you can also:
When you use the 3D-Sketcher, you manually place atoms in the model window, then refine the atom positions later (see Refining the conformation). Another builder tool, the Add Atom control panel, enables you to add atoms to the current model one at a time at specified coordinate positions. You can use it to build models and to add to existing models. Although less convenient to use than the Sketcher control panel, the Add Atom control panel allows precise control over the positions of atoms, which is particularly useful for duplicating published crystal structures or inorganic structures.
To access the Add Atom control panel, select the Build/Add Atom... menu item from the main control panel.
Several other atom properties can optionally be specified before placing an atom in the model window: hybridization, charge, occupancy, the atom name, and temperature factors (Specifying temperature factors)--see the on-screen help for details. Hybridization is also discussed under Changing the hybridization and adjusting the valence--the hybridization popup in the Add Atom control panel functions the same as in the Sketcher control panel.
After setting all desired parameters, click the ADD ATOM action button to place the atom in the model window. The most recently added atom can be deleted by clicking the UNDO button.
Adding templates (groups of atoms)
The efficiency of the building process can often be improved by adding groups of atoms ("templates") rather than single atoms. You may start sketching a new model by placing a template in the model window, or you can add templates to an existing model.
| This section refers to using the Sketcher control panel (The 3D-Sketcher), unless otherwise indicated. |
Selecting templates
The current template (the one that is used if you click in the model window while the template tool is active) is printed on the Sketcher control panel, just to the right of the template tool, for example:![]()
A specific example of using the file browser to find and load a template is demonstrated under The Templates... button looks like:.
Once you find the desired template file, load it by:
Once the desired template is chosen (see Selecting templates), click the template tool to activate it, if it is not already active. The cursor is now shaped like a T when it is over the model window. You can use the template to start building a model or to add a group to an existing model:
To save a template if you don't have the Analog Builder, follow these steps:
1. Construct the template model, making sure you are satisfied with its final structure and conformation (this section).
3. Replace this hydrogen with a dummy element of type X
(Changing the element type).
4. Save the model in .msi format (Saving model structure files).
You should now be able to use your template just as you do any other template, except that the Sketcher Template control panel does not find it automatically unless you were able to save it in the default Cerius2-Models/templates directory.
Fusing and connecting fragments
The Sketcher control panel includes tools for fusing or connecting two entities to form one model.
Fusing two fragments (in the model window) means to join them so as to delete a heavy (non-hydrogen) atom. For example, two benzenes could be fused to form naphthalene, or many cyclohexanes and cyclopentanes could be fused in building buckminsterfullerene.
![]()
Connecting two fragments means to join them by a bond (deleting only hydrogens as necessary). For example, two benzenes could be connected to form biphenyl or connected twice to form biphenylene.
![]()
| You can also connect fragments with the Sketch with tool (see What next?). |
| This section refers to using the Sketcher control panel (The 3D-Sketcher), unless otherwise indicated. |
You may add hydrogens to your model at any time and in several ways.
If any of the automatic tools give the wrong number of hydrogens at some atom(s), you can change their hybridization state (Changing the hybridization and adjusting the valence), formal charge (Changing the formal charge), and/or the bond type between that atom and another heavy atom (Changing the bond type) and then adjust the hydrogens again.
Why read this section
![]()
Selecting atoms and groups of atoms
You need to select one or more atoms when you want to perform some function (e.g., editing, labeling, changing the display style) on only certain atoms.
The controls
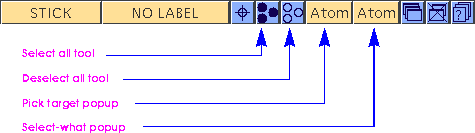
The Edit pulldown menu and the toolbar in the Visualizer's main control panel offer several ways of selecting one or more atoms. There are also four atom selection tools in the toolbar:
Several ways of selecting individual atoms are demonstrated under Selecting atoms.
You can also select atoms in several models simultaneously (Selecting atoms in several models).
Selecting and deselecting individual atoms
![]()
| For some functions, selecting all atoms has the same effect as selecting none (e.g., changing the display style, Model display style, or applying labels, Labels). Other functions (e.g., deleting all atoms, Deleting atoms and bonds) require that all atoms actually be selected. |
Managing and displaying multiple models is documented in Managing Models and Sessions.
Selecting atoms works differently, depending on how the models are displayed (see Controlling model visibility and the display mode):
A single pick in the model window (by the same selection process as described under Selecting and deselecting individual atoms) can act on different pick targets. A single click can pick a single atom, all atoms in one residue, all atoms in one chain, or all atoms in one model. Similarly, using a selection rectangle can pick only the atoms within the rectangle, or all residues, chains, or models that have at least one atom within the rectangle.
To change the default behavior (i.e., picking individual atoms), change the pick target popup on the toolbar (see The controls) from Atom to Residue, Chain, or Model. Residues and chains are not present in all models, but may be defined in other Cerius2 modules, which are documented separately.
When you pick a target, other items (in addition to the target) might also be selected, depending on your selection criteria.
Depending on the setting of the pick target popup, you can also select all residues of the same Type or Identifier or all chains of the same Name. The default is to select all atoms in a residue, chain, or model.
In addition to the selection criteria, you can restrict or expand the selection scope; for example, whether atoms are selected in the current model or all models, etc. To change the default selection scope (i.e., within the current model, use the Atom Selection control panel (see Additional selection criteria).
Not all combinations of pick target, selection criterion, and selection scope make sense or are allowed. The supported combinations are shown in Table 2.
| 1
Default value. 2 Or in all visible models or in selected models.
|
Select the Edit/Selection... menu item to access the Atom Selection control panel. This control panel enables you to specify the Pick target, Select atom(s) which are, and Within criteria when the mouse is used for selection (see Selecting according to properties).
In addition, more selection criteria are available. These functions are not linked to mouse activity in the model window, but occur when an icon or action button in the control panel is clicked.
Please see the on-screen help for details on the functioning of each control in the Atom Selection control panel.
Defining and selecting groups
Defining groups
You can define any collection of atoms as a "group" and then perform group-based selections.
You can select groups by clicking the Select objects action button in the Groups control panel or the Select Group button in the Atom Selection control panel.
Customized atom selection
You can set up sophisticated and/or multiple selection criteria and/or obtain even more precise control over what is selected when you pick an atom.
Select the Edit/Selection... menu item to access the Atom Selection control panel. Click the Custom Selection... pushbutton to access the Customized Atom Selection control panel.
To set up customized selection criteria, you can pick properties from the popups in the Customized Atom Selection control panel and enter additional subcriteria in the associated entry boxes.
To enter information into the subcriteria boxes, you of course need to know what an atom's properties are.
Please see the on-screen help for the precise syntax for entering subcriteria and for details on the functioning of each control in the Customized Atom Selection control panel. The help that is accessed with the ? buttons on this control panel is context-sensitive; that is, what help is displayed depends on what the associated popups are set to. The cumulative criteria are displayed in the Object Reference entry box as they are built up with the other controls in this control panel. The context of the Object Reference entry box may be edited.
Why read this section
![]()
Basic editing
Related information
Basic building tasks are presented under Basic building.
Tasks that may be less often or less typically performed, such as editing atom attributes and changing defaults and preferences, are covered in Advanced building and editing. Displaying labels and other information about atoms in a model is presented in Labels.
You need to know how to select atoms in your model (Selecting atoms and groups of atoms), so you can edit them. You also of course need to know how to build a model (Basic building) and/or load a model from a file (Loading model structure files).
You may want to review the basics of how the controls in the Sketcher control panel work (see How it works).
| This entire section refers to using the Sketcher control panel (The 3D-Sketcher), unless otherwise indicated. Most of its editing functions work with both nonperiodic and periodic (Other builders) systems. |
Changing atoms and their bonding types
This section includes:
To change the element type of an existing atom in the model window, you use the Edit Element tool.
Specifying the element type
The edit element type (i.e., the element that you want the atom to become) is listed in an entry box to the right of the Edit Element tool:
![]()
Changing the default element type automatically activates the Edit Element tool. If the edit element type is already correct, you can activate the Edit Element tool by clicking it.
If you want to edit both the element type and other properties simultaneously, select the Build/Edit Atoms... menu item. This gives access to the Edit Selected Atoms control panel.
Please see the on-screen help for details on the functioning of each control in the Edit Selected Atoms control panel.
Changing the formal charge
![]()
Choose AUTO or the desired value of the formal charge from the popup to the right of the Edit Formal Charge tool.
Changing the formal charge together with other properties
If you want to edit both the formal charge and other properties simultaneously, select the Build/Edit Atoms... menu item. This gives access to the Edit Selected Atoms control panel.
Please see the on-screen help for details on the functioning of each control in the Edit Selected Atoms control panel.
Changing the bond type
![]()
Choose SINGLE, DOUBLE, TRIPLE, or RESONANT from the popup to the right of the Edit Bond tool.
| You can also change the bond type while in sketch mode (see While in sketch mode, you can also:). |
Additional controls over bonding
If you want to edit the bond type and also change how bonds are calculated (see Editing element and bond attributes), select the Build/Edit Bonds... menu item. This gives access to the Edit Connectivity and Bonding control panel.
Please see the on-screen help for details on the functioning of each control in the Edit Connectivity and Bonding control panel. (Setting the bond calculation preferences is discussed under Bond properties.)
Changing the hybridization and adjusting the valence
Generally, Cerius2 automatically determines what the hybridization should be. However, you can specify the hybridization manually.![]()
| You can label atoms (Labels) according to their hybridization. |
Choose the desired type of hybridization from the popup to the right of the Edit Hybrid tool. AUTO means to allow Cerius2 to automatically determine the hybridization; the specific types listed in the popup are self-explanatory; OTHER should be used for high-coordination centers not specifically included in the list of choices. (Setting the hybridization to OTHER where appropriate allows the cleaning function (Refining the conformation) to work optimally.)
Changing the default hybridization type automatically activates the Edit Hybrid tool. If the hybridization type option is already correct, you can activate the Edit Hybrid tool by clicking it.
You can change the hybridization before adding an atom by using the Add Atom control panel (Precise positioning of atoms during building). You can also edit the hybridization of existing atoms (and other properties simultaneously) with the Edit Selected Atoms control panel (Changing the element type together with other properties).
Editing the structure
This section includes:
To delete an atom and all bonds leading to it, use the Delete Atom tool in the Sketcher control panel.
![]()
To delete a bond between two atoms without deleting any atoms, use the Delete Bond tool.
![]()
You can also copy or move parts of models, or entire models, from one model space to another (see Moving models between model spaces).
Examining and changing bond lengths
| Cleaning a structure (Refining the conformation) automatically optimizes all bond lengths. You can change bond lengths before cleaning if you want to bias the final structure. However, if you want a different specific bond length than that determined automatically, change it after using the clean function, do not use cleaning, clean only selected atoms (Control of the model-cleaning function), or change the default properties used for setting bond lengths (Bond properties). |
Changing a bond length manually
To change the length of a bond manually, select the Stretch tool in the Sketcher control panel.
![]()
To change the length of a bond precisely, select the Move/Bond Geometry... menu item to access the Bond Geometry control panel.
Please see the on-screen help for details on using the controls in the Bond Geometry control panel to measure and change bond lengths, valence angles, and torsions and to specify which atoms are moved in the process.
You can change the defaults that Cerius2 uses in calculating bonds. To change the default radii of atoms, please see Element properties. For covalent bond calculation preferences, see Bond properties. For setting the hydrogen bond length criterion, see Hydrogen bonds.
| Cleaning a structure (Refining the conformation) automatically optimizes all valence angles. You can change angles before cleaning if you want to bias the final structure. However, if you want a different specific angle than that determined automatically, change it after using the clean function, clean only selected atoms (Control of the model-cleaning function). or do not use cleaning. |
To change a valence angle manually, select the Angle tool in the Sketcher control panel.
![]()
To change an angle precisely, select the Move/Bond Geometry... menu item to access the Bond Geometry control panel.
Please see the on-screen help for details on using the controls in the Bond Geometry control panel to measure and change valence angles, bond lengths, and torsions and to specify which atoms are moved in the process.
You can implicitly control default valence angles through setting the hybridization state of the atoms involved (Changing the hybridization and adjusting the valence). For setting the hydrogen bond angle criterion, please see Hydrogen bonds.
Examining and changing torsions
| Cleaning a structure (Refining the conformation) automatically optimizes all torsion angles. You can change torsions before cleaning if you want to bias the final structure. However, if you want a different specific torsion angle than that determined automatically, change it after using the clean function, clean only selected atoms (Control of the model-cleaning function), or do not use cleaning. |
To change a torsion angle manually, select the Twist tool in the Sketcher control panel.
![]()
To change a torsion angle precisely, select the Move/Bond Geometry... menu item to access the Bond Geometry control panel.
Please see the on-screen help for details on using the controls in the Bond Geometry control panel to measure and change torsions, bond lengths, and valence angles and to specify which atoms are moved in the process.
You can implicitly control default torsion angles through setting the hybridization state of the atoms involved (Changing the hybridization and adjusting the valence).
Inverting about an atomic center
![]()
| Inversions cannot be done for centers that are part of complex ring structures, i.e., for those in which one fragment makes more than two attachments to the selected atomic center. |
You can examine inversions with the Measurements control panel (Inversions).
Translating and rotating atoms
Translating atoms manually
Tables of mouse button and keyboard combinations are included in Mouse and Keyboard Actions.
To translate selected atoms a precise distance in a specified direction, select the Move/Atoms Translate... menu item to access the Translate Atoms control panel.
Please see the on-screen help for details on the functioning of each control in the Translate Atoms control panel.
Tables of mouse button and keyboard combinations are included in Mouse and Keyboard Actions.
To rotate selected atoms by a precise angle around a specified rotation axis and center, select the Move/Atoms Rotate... menu item to access the Rotate Atoms control panel.
Please see the on-screen help for details on the functioning of each control in the Rotate Atoms control panel. You can rotate the selected atom(s) by clicking and dragging a slider or a dial or by entering the angle in the associated entry box.
Aligning and positioning atoms
Aligning atoms
To accurately align selected atoms with respect to a coordinate system, select the Move/Atoms Align... menu item to access the Align Atoms control panel. You specify one intra-model vector that is to be aligned with one coordinate axis and another intra-model vector to be aligned with a coordinate plane that includes that axis. You also specify the point about which the selected atoms are rotated.
Please see the on-screen help for details on the functioning of each control in the Translate Atoms control panel.
To accurately position selected atoms in space, select the Move/Atoms Position... menu item to access the Position Atoms control panel. You specify both a reference point and a destination point, then the selected atoms are moved in a way that maintains their spatial relationship with the reference point (i.e., the reference point is moved together with the atoms).
Please see the on-screen help for details on the functioning of each control in the Position Atoms control panel. (You can position atoms while adding them to a model, with the Add Atom control panel, see Precise positioning of atoms during building.)
Refining the conformation
Cleaning the structure
The model structure as initially drawn is usually not chemically reasonable. Thus, it needs to be "cleaned" (i.e., its structure needs to be optimized). Initial optimization is often done with the CLEAN button in the Sketcher control panel. Simply click the CLEAN button and keep the left mouse button depressed until the structure in the model window stops changing. (You can adjust how the clean function works--please see Control of the model-cleaning function.)
You may adjust the structure as desired after you clean it (e.g., Examining and changing bond lengths). However, a better approach may be to apply constraints before additional optimization or to apply symmetry (next paragraph).
If you want an even more chemically correct structure than the clean function is capable of, you may want to use a forcefield-based minimizer such as the OFF Minimizer or optimize the structure to an energy minimum or a transition state with one of the quantum applications. You can also use the quantum applications to enforce space-group symmetry. Documentation for these modules is supplied separately.
Why read this section
![]()
Advanced building and editing
Related information
Basic building tasks are presented under Basic building and basic editing tasks under Basic editing.
Displaying labels and other information about atoms in a model is presented under Labels.
Saving nondefault settings for later use is described under Working with Cerius2 sessions.
You need to know how to select atoms in your model (Selecting atoms and groups of atoms), so you can edit them. You also of course need to know how to build (Basic building) and edit (Basic editing) a model and/or load a model from a file (Loading model structure files).
You may want to review the basics of how the controls in the Sketcher control panel work (see How it works) or how controls work in general (The Cerius2 Interface).
Editing element and bond attributes
Cerius2 allows you to edit the default properties of elements (Element properties), change how bonds are determined (Bond properties), and adjust the criteria for determining hydrogen bonds (Hydrogen bonds).
Changing the properties of individual selected atoms (i.e., not necessarily of all atoms of a given element type) is covered under Changing atoms and their bonding types. Setting temperature factors is discussed under Temperature factors.
You should have already worked with the types of models and the various application modules that you typically use, before deciding whether and how much you should change the default behavior of Cerius2. And of course there is no substitute for chemical knowledge and intuition. This section is for experienced Cerius2 users.
Element properties
What element properties
can be edited
Element properties are stored within Cerius2 and used for calculations and display attributes. These properties include mass, van der Waals radius, atomic radius, covalent radius, metallic radius, ionicity, hydrogen bonding nature, and display color. Cerius2 "elements" include chemically real elements, "mixture" elements, and "dummy" elements.
Select the Build/Element Defaults... menu item to access the Edit Elements control panel. Changing element defaults changes the properties for atoms of that element type that already exist in the model, as well as for all atoms added to models later in the current Cerius2 session.
Please see the on-screen help for details on the functioning of each control in the Edit Elements control panel.
Mixture elements are special Cerius2 elements (with atomic symbols T, TT, M, and MM) whose properties you can specify as proportions of those of up to eight real elements. Such a mixture can be useful, for example, in studying disordered structures (Introducing substitutional disorder).
Mixture elements are defined using the Element Mixtures control panel, which you access by clicking the Mixtures... pushbutton on the Edit Elements control panel. You can also examine the compositions of predefined mixture elements with this control panel.
You can also edit properties of individual selected atoms (rather than of all existing and future atoms of a given element type): the element type, hybridization state (Changing the hybridization and adjusting the valence), charge, formal charge, mass, occupancy, the atom name, and temperature factors (Specifying temperature factors). To do so, select the Build/Edit Atoms... menu item to access to the Edit Selected Atoms control panel.
Please see the on-screen help for details on the functioning of each control in the Edit Selected Atoms control panel.
Bond properties
Bonds can be drawn and then optimized automatically (Refining the conformation). Alternatively, unbonded atoms can be placed in the model window (see Precise positioning of atoms during building) and then bonds can be calculated for all or selected atoms. In either event, the calculated bond lengths are based on distance and other criteria. For cleaning, Cerius2 uses these criteria in deciding what is the optimum bond length. For placing bonds, Cerius2 uses them to decide which atom pairs to join by bonds.
Elementary bond editing with the Sketcher and the Edit Connectivity and Bonding control panels is presented under Changing atoms and their bonding types and Editing the structure. These tasks include deleting bonds, changing the bond type and order, and making bonds between selected atoms.
For more advanced bond editing tasks or for performing several elementary tasks simultaneously, select the Build/Edit Bonds... menu item to access the Edit Connectivity and Bonding control panel.
Please see the on-screen help for details on the functioning of each control in the Edit Connectivity and Bonding control panel.
To define the criteria by which bonds are calculated, click the Preferences... pushbutton to the right of the Calculate connectivity check box in the Edit Connectivity and Bonding control panel to access the Bond Calculation Preferences control panel.
You can also define a Bonding Tolerance (effectively the proportion of the relevant atomic radius to be used in looking for contacts). For example, if the tolerance factor is 1.5 and the ideal bond length is 1.4 Å, a bond is created if the two atoms are within 2.1 Å (1.4 x 1.5) of one another.
Please see the on-screen help for details on the functioning of each control in the Bond Calculation Preferences control panel.
To specify whether Cerius2 should determine the bond types automatically by valence satisfaction, check the Calculate bond types check box. To specify how this algorithm works, click the Preferences... pushbutton next to the Calculate bond types check box in the Edit Connectivity and Bonding control panel to access the Bond Type Calculation Prefs control panel.
Please see the on-screen help for details on the functioning of each control in the Bond Type Calculation Prefs control panel.
To request bond calculation between all atom pairs that meet the criteria, click the CALCULATE pushbutton in the Edit Connectivity and Bonding control panel. Bonds are added or deleted so that they meet the criteria. Subsequent use of the cleaning function (Refining the conformation) also uses the radii set with the Bond Calculation Preferences or Edit Elements control panel.
Hydrogen bonds
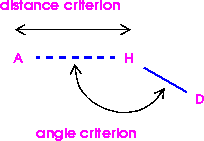
All or selected hydrogen bonds can be edited and deleted. Hydrogen bonds can also be calculated for all or selected atoms, based on distance and angle criteria.
When Cerius2 decides which atom pairs to join by hydrogen bonds, it uses these criteria (Figure 1):
|
Select the Build/Edit H-Bonds... menu item to access the Edit Hydrogen Bonding control panel.
Please see the on-screen help for details on the functioning of each control in the Edit Hydrogen Bonding control panel.
To define the criteria by which hydrogen bonds are calculated, click the Preferences... pushbutton in the Edit Hydrogen Bonding control panel, to access the H-Bond Preferences control panel.
When these criteria are altered, any existing hydrogen bonds that no longer meet the criteria are deleted when you click the CALCULATE pushbutton in the Edit Hydrogen Bonding control panel. Hydrogen bond recalculation occurs automatically if the Enable automatic recalculation check box in the Edit Hydrogen Bonding control panel is checked.
Please see the on-screen help for details on the functioning of each control in the H-Bond Calculation Preferences control panel.
To request hydrogen bond calculation between all or selected atom pairs that meet the criteria, click the CALCULATE pushbutton in the Edit Hydrogen Bonding control panel.
Introducing substitutional disorder
Uses of substitutional disorder
Substitutional disorder can be used whenever two elements need to be randomly replaced in a structure. For example, substitutional disorder is particularly useful in dealing with zeolites where you may want to randomly replace silicon atoms with aluminum.
Substitutional disorder randomly replaces a fraction of the atoms of one element type with atoms of another element type. Substitutional disorder can be applied to the whole model or to only a selected portion of the model. A target ratio is used to set the fraction of the atoms to be replaced.
| To apply disorder to symmetric crystal structures, first convert the structure to a primitive superlattice (as described in the Cerius2 Builders documentation) and then perform substitution. |
Performing substitutional disorder
Please see the on-screen help for details on the functioning of each control in the Disorder control panel. After the substitution is performed, information on the number of atoms replaced, etc., is shown in the text window.
Introducing positional disorder
Uses of positional disorder
Positional disorder is a convenient way to move a model away from a local energy minimum. For example, you can apply positional disorder before cleaning a model (see Refining the conformation) or before performing an energy minimization (see the documentation for the Minimizer module in Cerius2 Simulation Tools).
Positional disorder randomizes atomic positions--atoms are moved by the specified displacement in a random direction. Positional disorder can be applied to any model type. However, if an atom in a periodic structure is in a special symmetry position, it is not moved from that position, although it can be displaced in allowed directions. For example, if an atom lies on a symmetry axis or plane, it (and all its symmetry copies) is moved only along that axis or within the plane, respectively. The distance the atom is moved is the component of the random displacement vector that lies on the symmetry axis or in the symmetry plane.
Please see the on-screen help for details on the functioning of each control in the Disorder control panel.
Temperature factors
Finding information
Related information
Representing the thermal motions of atoms graphically is discussed under Thermal ellipsoids.
Temperature factors and atomic vibrations
Atomic vibrations are described in the lowest order by their temperature factors. If the deviation of an atom from its equilibrium position is r = (x,y,z), the vibration is characterized by the matrix:
Eq. 1
Where 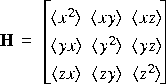
![]() xy
xy![]() represents the average over many vibrations.
represents the average over many vibrations.
H is symmetric and contains six independent quantities, the anisotropic temperature factors Uij. If the atomic vibration is isotropic, then H reduces to the form
![]() x2
x2![]() I and requires just one number (the isotropic temperature factor Uiso) to describe it:
I and requires just one number (the isotropic temperature factor Uiso) to describe it:
Eq. 2
Effects of vibration on
X-ray scattering
![]()
Temperature factors are commonly used to describe the effect of vibration on X-ray, electron, and neutron scattering. For the first-order amplitude of scattering from an atom at q = ha* + kb* + lc* in reciprocal space, the scattering amplitude is multiplied by:
Eq. 3
Conventions
![]()
Several different though equivalent ways of describing the temperature factors exist. (Cerius2 can read temperature factors in several formats, see Loading and saving temperature factors.) Sometimes the vibration is described in terms of a crystal lattice coordinate system (a,b,c) rather than a Cartesian one.
Cartesian and crystal coordinate systems
If an atom has coordinates (x,y,z) with respect to the Cartesian axial system and (u,v,w) with respect to the lattice system so that:
Eq. 4
then (x,y,z) and (u,v,w) can be interconverted by multiplying by a matrix A where (x,y,z)T = A(u,v,w)T.![]()
| Form |
Relationship to H or | Scattering amplitude multiplied by |
|---|---|---|
| Uiso |
|
|
| Biso |
|
|
| Uij 1 |
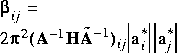
|
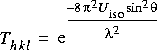
|
| Bij 2 |
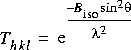
|
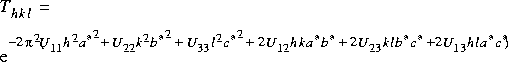
|
|
|
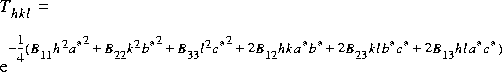
|
|
| 1
a* = (b ^ c)/(a· b ^ c), etc. 2 For
|
Temperature factors in Cerius2
Internally, Cerius2 stores the atomic vibration matrix H. Cerius2 stores isotropic and anisotropic temperature factors for every atom. You decide which of the two types to use for calculation or display.
Specifying temperature factors
Temperature factors can be specified when you add new atoms. Select the Build/Add Atom... menu item to access the Add Atom control panel. In this control panel, click the Temperature Factors... pushbutton to access the Added Atom Temperature Factors control panel.
![]() ij for anisotropic temperature factors; see Table 3).
ij for anisotropic temperature factors; see Table 3).
U11 U12 U13The selected specifications are applied when you add the atom with the Add Atom control panel (Precise positioning of atoms during building).
U22 U23
U33
If you specify the anisotropic temperature factors from the command line, you should enter them in the order (1,1) (2,2) (3,3) (1,2) (2,3) (1,3).
Please see the on-screen help for details on the functioning of each control in the Added Atom Temperature Factors control panel.
Editing temperature factors
The temperature factors of existing atoms can be edited. Select the Build/Edit Atoms... menu item to access the Edit Selected Atoms control panel. In this control panel, click the Temperature Factors... pushbutton to access the Edit Temperature Factors control panel. With this control panel you can:
U11 U12 U13
U22 U23
U33
If you specify the anisotropic temperature factors from the command line, you should enter them in the order (1,1) (2,2) (3,3) (1,2) (2,3) (1,3).
Please see the on-screen help for details on the functioning of each control in the Added Atom Temperature Factors control panel.
Loading and saving temperature factors
As mentioned (Conventions and Table 3), several formats exist for describing temperature factors. When temperature factors are loaded from or saved in structure files, the factors are automatically converted to and from the Cerius2 format.
The MSI, SHELX, CIF, ICSD, DBWS, and MolEN structure file formats can all be used to store isotropic and anisotropic temperature factors (please see File Formats for more details).
Isotropic temperature factors (but not anisotropic factors) are stored in both .xtl and .car/.mdf files.
Control of the model-cleaning function
The default mode of operation of the cleaning function is as described under Refining the conformation. However, you may want it to function differently.
You can specify the mode of operation of the cleaning function, the number of cycles performed (for one-shot cleaning), which atoms are to be cleaned, and which atoms are to be taken into account during the clean.
Click the Preferences... pushbutton near the bottom of the Sketcher control panel to access the Cleaner Controls control panel.
Please see the on-screen help for details on the functioning of each control in the Cleaner Controls control panel.
Tools designed to build more specialized models are available as separate modules and are accessed with the deck of cards menu. These include the Analog Builder, Crystal Builder, Surface Builder, Interface Builder, Polymer Builder, and Amorphous Builder. These modules are described in Cerius2 Builders.![]()
Other builders